
{kind=link}
从炎症调节因子到肿瘤抑制因子——A20的特点及其在淋巴细胞肿瘤发生中的作用
引用本文
李扬秋, 杨力建, 陈少华, 李萡. 从炎症调节因子到肿瘤抑制因子——A20的特点及其在淋巴细胞肿瘤发生中的作用. 循证医学,2011,11(1): 54-59
LI Yang-qiu, YANG Li-jian, CHEN Shao-hua, LI Bo. From Inflammation Regulator to Tumor Suppressor——The Feature of A20 and Its Role in the Initiation of Lymphocytic Malignancy. Journal of Evidence-based Medicine, 2011,11(1): 54-59
Permissions
LI Yang-qiu, YANG Li-jian, CHEN Shao-hua, LI Bo. From Inflammation Regulator to Tumor Suppressor——The Feature of A20 and Its Role in the Initiation of Lymphocytic Malignancy. Journal of Evidence-based Medicine, 2011,11(1): 54-59
Copyright©2011, 《循证医学》杂志 版权所有
从炎症调节因子到肿瘤抑制因子——A20的特点及其在淋巴细胞肿瘤发生中的作用
作者简介:李扬秋(1962-), 女, 广东汕头人, 医学博士, 研究员, 博士研究生导师, 主要从事血液肿瘤免疫学研究。
摘要
A20是炎症信号传导的中心调节因子—NF-κB抑制因子,而近年研究发现A20更重要的功能是作为一个肿瘤抑制因子,其缺失在淋巴细胞肿瘤的发生中有重要的作用。综述近年所报道的A20生物学功能、分子机制及其在淋巴细胞肿瘤中调节作用的研究进展。
关键词:
淋巴细胞肿瘤; A20; 肿瘤抑制因子
中图分类号:R733.4
文献标识码:A
文章编号:1671-5144(2011)01-0054-06
From Inflammation Regulator to Tumor Suppressor——The Feature of A20 and Its Role in the Initiation of Lymphocytic Malignancy
Abstract
A20 was originally identified as a key regulator of inflammation signalling pathways, a NF-κB inhibitor. Recently, A20 has also been proposed to function as a tumor suppressor. Lacking A20 is involved in tumorigenesis in several human B-cell lymphomas. In this review, current knowledge concerning the feature of A20 biological activities, as well as the underlying molecular mechanisms, and the regulating role of A20 in lymphocytic malignancies is summarized.
Key words :
lymphocytic malignancies; A20; tumor suppressor
近年来逐步认识到非可控性炎症在肿瘤微环境形成、导致细胞恶性转化中起关键性作用[1]。炎症性疾病如类风湿性关节炎, 始发于促炎相关细胞因子和趋化因子、炎症启动酶以及免疫受体的异常表达和刺激, 在这些过程中, 转录因子NF-κ B起关键性的作用。目前已明确多数炎症的调控因子均为NF-κ B依赖性。同时, NF-κ B也诱导了系列抗凋亡基因的表达, 扮演了抗凋亡的重要角色。可以想象, 某些控制NF-κ B规律性和限制性表达的调控因子在炎症甚至细胞恶性转化过程中具有重要的作用。A20作为一个NF-κ B的负调控因子, 其异常必然影响到炎症的发生和转变, 其调控细胞凋亡的机制失常必然与肿瘤的发生发展密切相关[2, 3, 4]。
1 A20的结构和表达特点
首先, A20是在肿瘤坏死因子α (tumor necrosis factor α , TNF-α )诱导内皮细胞反应中所检测到的一个诱导性表达蛋白, 因此也被称为TNF-α 诱导蛋白3(TNFAIP3)[5]。A20基因定位于染色体6q23.3, 其cDNA序列为4 440 bp, 其开放读码框为2 370 bp, 编码790氨基酸的90 kDa蛋白。A20蛋白在羧基末端含有7个Cys2-Cys2锌指(zinc finger, ZnF)基序, 是一种新类型的锌指蛋白, 这些锌指基序是NF-κ B信号传导作用所必须的。不同锌指的作用也逐步被明确, 如A20的第四锌指(ZnF4)的作用有一定特殊性, 它不能直接与E2连接酶结合, 只能结合单个泛素或K63-连接的泛素链。而A20的氨基端则含有一个基序, 称为卵巢肿瘤基序(ovarian tumor domain, OTU), 具有去泛素化蛋白酶活性作用[6, 7, 8]。
除了在TNF-α 诱导内皮细胞中首先发现A20以外, 后期的研究发现, 多种类型细胞和多种刺激源(包括B细胞表面受体CD40、EBV-LMP1等)都可以诱导A20的表达。A20的构成性表达可见于不成熟的双阳性和单阳性胸腺细胞, 随着单阳性细胞成熟, A20的表达水平下降, 外周静息T细胞也表达一定水平的A20, 但随着T细胞的活化其表达水平下调。此外, A20也表达于多种单核细胞株中, 在中枢神经系统、脊髓、肝脏、肺和心脏也有低水平的表达[2]。
A20基因缺失、突变和多态性在自身免疫性疾病和淋巴细胞肿瘤中已有报道, 尤其在自身免疫性疾病中, 某些基因位点多态性与疾病易感性相关。如单核苷酸多态性(single nucleotide polymorphism, SNPs)rs5029939 G与系统性硬化症的易感性高度相关[9]。
2 A20的生物学功能
A20是一个具有双重功能的泛素编辑酶。最早发现的A20的功能是作为TNF诱导凋亡的抑制因子。A20在一些细胞株中稳定高表达显示出对TNF诱导凋亡的抵抗作用, 从而认为A20是一个抗凋亡分子。但该作用并非是一个普遍现象, 如在MCF7(人乳腺癌细胞株)中, A20则不具有抗凋亡作用。此外, 另一个更重要和更为普遍的发现是A20具有重要的NF-κ B抑制因子的功能, 即限制NF-κ B诱导的抗凋亡作用。A20双重性功能的机制目前尚不明确[2]。后期多数研究关注其作为NF-κ B抑制因子的作用, 这一作用涉及了炎症和免疫系统多种相关的病理生理过程、免疫应答变化和自身免疫性疾病等。而近期的更多研究则提示, A20的重要功能实际上是作为肿瘤抑制因子, 其缺失与淋巴细胞肿瘤的发生关系密切[7]。
2.1 NF-κ B抑制因子
对A20启动子的克隆和特征分析发现其具有NF-κ B转录因子的两个识别序列的κ B元件。由于NF-κ B是TNF-α 活化炎症反应的关键调节因子, A20自身的表达又受到NF-κ B的控制, 因此, 提示A20参与了负调控环而延缓了TNF-α 活化炎症反应。随后的研究显示其他刺激如佛波醇酯以及病毒蛋白如HTLV1-Tax和EBV-LMP1诱导B细胞表面受体CD40活化过程中, 也诱导了A20转录。超表达A20可阻断不同类型细胞中TNF、IL-1和LPS等诱导的NF-κ B的活化。在平滑肌细胞体外培养研究中, A20同样通过影响NF-κ B活化而发挥抗增殖作用, 所有这些结果都提示A20具有延缓促炎信号传导的作用, 它主要通过干预调节和活化NF-κ B的上游信号通路而负调控NF-κ B依赖的基因的表达[2, 7]。
2.2 免疫调节因子
NF-κ B在淋巴细胞的活化、存活和增殖的调节中起关键作用, A20作为多个NF-κ B活化信号通路的重要调节因子, 同样调节免疫应答中NF-κ B活化信号传导过程。总的来说, A20在调节免疫应答中, 预防不同外源刺激反应时NF-κ B过度活化中起关键作用[10]。
A20对天然免疫和获得性免疫细胞均具有调节作用。对于T细胞和B细胞的调节主要是通过CARMA1-Bcl-10-MALT1(CBM)复合物信号通路而发挥作用。在T细胞或B细胞相应的第一信号系统TCR或BCR活化后, 分别导致了其下游丝氨酸-苏氨酸激酶PKC-θ 和PKC-β 的活化, 通过系列链锁的募集反应, 引起了CARMA1-Bcl-10-MALT1(CBM)复合物的形成, 而CBM复合物将TCR/BCR信号网络连接到经典的Iκ B激酶(IKK)/NF-κ B通路上[11]。在T细胞中, A20通过调节IKK/NF-κ B对TCR/CD28共刺激反应的强度和持续时间而负调控TCR信号传导通路[12]。A20缺失小鼠模型中B细胞活化的阈值降低, 增加促炎细胞因子的表达(尤其是IL-6), 引起了进展性炎症反应, 包括了粒细胞、效应性T细胞和调节性T细胞扩增, 最终小鼠可发展为自身免疫性综合征[13]。
此外, A20是Toll样受体信号传导通路的负调控因子, 它在控制DC的成熟、细胞因子的产生、免疫刺激能力等方面都有重要作用。A20沉默的DC显示了自发性和增强共刺激分子和促炎细胞因子的表达, 并具有抑制Treg细胞、增强肿瘤浸润细胞毒T细胞和T辅助细胞应答的效应, 可以认为A20作为一个抗原提呈参与者控制了抗肿瘤免疫应答[14]。
2.3 肿瘤抑制因子
基于慢性炎症与肿瘤发生之间的关联性, 不难想象A20突变性失活导致NF-κ B表达失控与肿瘤发生的相关性。近期在多种淋巴瘤中发现了由于缺失、启动子甲基化、移码突变和/或点突变等导致A20失活的现象, 且常见A20两个等位基因同时受累, 这是肿瘤抑制因子常见的特点。然而, 另一种完全不同的特点则是A20高表达于未分化鼻咽肿瘤和低分化头颈部鳞状细胞癌中, 而正常鳞状细胞和高分化头颈部鳞状细胞癌并不表达A20, A20也表达于雌激素受体和孕激素受体阴性乳腺癌细胞中, 并且与预后不良相关。A20的这一特点可能与其抗凋亡作用相关, 提示A20可能参与这些肿瘤的发病机制[2, 7, 15]。
在一个A20等位基因失活的淋巴瘤细胞株(KM-H2)中, 导入野生型A20后, 细胞的增殖受到抑制, 并诱导细胞发生凋亡, 在此过程中伴有NF-κ B活性下调[10]。相反, 当导入突变型A20后, 则不能发挥相应的作用。免疫荧光染色可见A20导入后, P50在细胞浆中的定位发生改变, 证实了NF-κ B信号传导的A20依赖性阻断反应。这更充分体现了A20作为肿瘤抑制因子的依据[16]。A20缺失的B细胞在免疫缺陷小鼠NOD/SCID/γ cnull(NOG)中可稳定形成肿瘤, 而肿瘤的发生、肿瘤细胞增殖和NF-κ B活性可以被A20的再表达所抑制, 均显示为A20再表达依赖性。
基于在多种上游刺激下A20对NF-κ B活性的负调节的生理学功能, 在淋巴瘤中这些发现提示A20功能的丧失导致NF-κ B信号传导失控参与了某些类型B细胞肿瘤的发病机制[10]。
3 A20结合蛋白及其作用机制
由于A20功能的双重性, 提示了其可能具有结合不同的靶分子而发挥不同的效应, 如A20与靶分子TXBP151结合所产生的作用是抗凋亡功能[2]。
3.1 A20结合蛋白
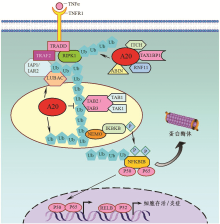
参与A20作用的蛋白主要是平行信号通路上的分子, 包括有TRAF家族系列分子如TNF受体相关因子TRAF2和TRAF6等, 以及ABIN系列分子和NEMO(IKBKB)等, 见图1[7, 15]。
3.2 A20抗凋亡作用机制
A20的抗凋亡作用机制仍不甚清楚。在某些类型细胞中, A20具有保护TNF等刺激诱导的细胞凋亡作用。A20首先结合和降解了凋亡信号调节激酶1(ASK1), 而抑制c-jun的氨基末端激酶(JNK)的活化作用, 从而阻断了凋亡的发生[17]。如在胶质瘤中, A20也显示了抗凋亡作用, 它通过降低细胞周期进程和减少p65/RelA的磷酸化相关机制而实现[18]。在T细胞白血病细胞株Jurkat细胞中, A20通过防止RIP1和TRADD募集形成TNF-R1复合物而抑制TNF诱导的细胞凋亡作用[15]。
3.3 抑制NF-κ B作用机制
A20含有去泛素酶和E3连接酶基序, 作为一个泛素编辑因子, 它通过灭活泛素化RIP1(受体相互作用蛋白1)而发挥TNFR1下游泛素编辑酶功能, 断开Lys-63(赖氨酸)-连接泛素链, 并增加泛素链作用底物的降解。该作用主要是由A20的羧基末端的锌指与E2或E3等酶接触而发挥效应。另一方面, A20与调节分子TAX1BP1(Tax 1结合蛋白1)联合, 与E2酶(Ubc13和UbcH5c)接触, 引起了它们的泛素化合蛋白酶体降解, 从而发挥了炎症信号传导通路的抑制作用[8, 19]。近年的系列研究提示, 在不同刺激作用下, A20与不同靶蛋白结合后对NF-κ B的抑制作用都是通过对不同类型的聚泛素链的解聚合作用即去泛素化而发挥其效应的[7]。
总的来说, A20的功能是通过多种机制来实现的, 它消除近端信号传导蛋白活化所需要的聚泛素链, 阻断E2和E3之间的联系, 在细胞活化信号传导早期限制了募集反应(如RIPK1)而形成信号通路复合物, 并进一步延长(如UbcH5c、UbE2N和TRAF2)在复合物中决定性蛋白的降解效应[7]。
3.4 A20抑制肿瘤作用的机制
A20在不同肿瘤发生中的作用机制有所不同, 在实体肿瘤如结肠腺癌动物模型中发现, 由于不同类型细胞中存在慢性NF-κ B活化, 它以协同作用形式促进了肿瘤发生:NF-κ B上调了肠细胞的原癌基因, 而肠道巨噬细胞产生NF-κ B诱导生长因子以旁分泌形式促进肠细胞增殖。而在淋巴瘤中, 生长因子信号的细胞来源及其特点并不完全清楚, 有可能是趋化到肿瘤部位的免疫细胞产生了生长因子, 或淋巴瘤细胞以自分泌形式产生生长因子和信号。例如, 来自KM-H2细胞条件培养上清可增加肿瘤细胞的NF-κ B信号传导(A20依赖性), 在加入细胞因子TNF-α 和淋巴毒素-α 的单克隆抗体阻断后, 则可抑制细胞生长和NF-κ B信号[7]。
在淋巴细胞肿瘤中, 多发现伴有A20缺失, 而A20缺失的B细胞对于多种刺激反应过强, 尤其是对CD40诱导信号显示了NF-κ B过度增加。同时, 对于Fas介导的细胞凋亡有抵抗作用, 该作用可能与NF-κ B依赖的抗凋亡蛋白如Bcl-x增加有关, 这是间接诱导肿瘤发生的相关机制[20]。在某些类型淋巴瘤中, A20的缺失是由于蛋白酶黏膜相关淋巴样组织淋巴瘤移位基因1(MALT1)的构成性活化而引起的, MALT1无论对于BCR还是TCR信号传导诱导的NF-κ B活化都是非常关键的, 它具有分解A20的作用, 其异常超表达直接抑制A20而引起NF-κ B过度表达, 导致NF-κ B的失控, 涉及MALT发病机制[7, 11]。
4 淋巴细胞肿瘤中A20失活情况
近年的研究已经普遍认识了A20的肿瘤抑制因子功能, 与其他肿瘤抑制基因类似, 其异常表达、缺失和突变必然与肿瘤的发生相关。由于A20主要通过负调控NF-κ B而涉及炎症的调节作用, 因此, 作为肿瘤抑制因子, A20可能存在一些组织特异性, 在B细胞肿瘤中, 首先发现了A20的异常变化现象。
4.1 淋巴瘤
由于在多种淋巴瘤中A20基因缺失、启动子甲基化、移码突变和/或点突变等导致A20的失活, 且常常累及A20两个等位基因, 体外基因导入提示恢复A20功能后可诱导肿瘤细胞凋亡和抑制增殖等, 认为A20在淋巴瘤中具有肿瘤抑制因子功能。所涉及的淋巴瘤包括有边缘区淋巴瘤(marginal zone lymphoma, MZL)、弥漫大B细胞淋巴瘤(diffuse large B-cell lymphomas, DLBCL)、套细胞淋巴瘤、黏膜相关组织(mucosal-associated lymphoid tissue, MALT)淋巴瘤、滤泡淋巴瘤(follicular lymphoma, FL)、原发性纵隔B细胞淋巴瘤、Burkitt淋巴瘤、NK细胞淋巴瘤、成人T细胞白血病/淋巴瘤和霍奇金淋巴瘤(Hodgkin’ s lymphoma, HL)等[7, 21]。
在各种淋巴瘤中, A20失活的发生率不等, 高者超过30%, 而在部分淋巴瘤中仅有个例发现A20异常改变。在一组大样本(238例)B细胞淋巴瘤的分析中发现, A20因为突变或缺失而失去活性在MALT淋巴瘤(18/87例, 21.8%)和结节硬化型HL(5/15例, 33.3%)中常见, 而在其它类型B细胞淋巴瘤中较为少见, 如在DLBCL中为5/64例, 在FL中为1/52例[10]。但在同期的另一个详细分析DLBCL样本的报道中, 则具体显示不同亚型DLBCL中A20异常改变的明显差异性:A20基因突变在活化B细胞(ABC)-DLBCL中发生率最高, 为9/37(24.3%), 在生发中心B细胞(GCB)-DLBCL中发生率最低, 为1/44(2.3%), 而在Non-GC/NC(未分型)-DLBCL中则为20%(4/20), 但A20失活的比例则更高, 两个等位基因A20缺失见于32%的ABC-DLBCL和34%的NON-GC/NC-DLBCL, 而单个等位基因缺失可见于23%的ABC-DLBCL和22%的NON-GC/NC-DLBCL中[16]。在神经糸统DLBCL— 原发性中枢神经系统淋巴瘤(PCNSL)中, A20基因突变仅见于3%病人(1/32), 在脊髓DLBCL中, A20突变为10%(1/10)[22]。而在不同MZL(包括结外MZL, 淋巴结MZL和脾脏MZL等)病人中也发现19%(6/32)存在A20突变和缺失情况[23]。在经典型HL(cHL)中, 发现A20突变的比例比较高, 达到44%(16/36), 且多涉及两个等位基因的改变, 引起A20失活, 也常见A20位点的染色体缺失[3]。
4.2 淋巴细胞白血病
在慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)中, 同样可以发现NF-κ B异常活化, 但两个研究报告的结果提示A20的突变和失活在CLL中少见, 一组选择性分析伴有6q23缺失的样本中, 仅发现25%伴有A20缺失, 仅有1例伴有A20突变。另一个报道检测55例CLL, 均未发现A20基因突变现象, 分析63例CLL, 均未发现A20基因启动子区甲基化状态, 因此, 提示在CLL中, A20缺失至少在大部分病人中, 并非是NF-κ B异常活化的原因, CLL的发生机制与B细胞淋巴瘤不同, 意味着尚有其他未发现的导致NF-κ B异常活化的原因。但一个有意义的发现是:伴有A20缺失者多为不伴有突变型IgH的预后差的CLL类型, 这也许是一个预后的生物标志物指标, 也可能是进一步研究其发病机制的一个新途径[24, 25]。
4.3 其他肿瘤
在其他实体肿瘤中, 有关A20异常改变的研究报道并不多, 新近报道比较有意义的结果是神经胶质瘤组织样本中A20的改变, 63.9%可以检测到A20的高表达, 并且与临床分期有关[26]。
5 基于A20的抗肿瘤和免疫治疗策略
在多种A20缺失的细胞株中, 包括DLBCL(SUDHL2、RC-K8)和cHL细胞株(L-1236)等, 导入野生型A20基因, 可显著减少NF-κ B的转录本, 使细胞获得凋亡的能力, 并抑制了细胞的增殖, 但是, 对于A20正常或没有发生NF-κ B构成性活化的细胞株, 导入A20并不能发挥作用[3, 16]。这一结果提供了A20作为基因治疗的靶向性依据, 因此, 针对A20缺失的肿瘤, 可以考虑将导入野生型A20基因作为一个抗肿瘤靶向治疗的策略。
A20通过调节DC而抑制Treg细胞的效应, 同样提供了一个克服抗原特异免疫过程中Treg细胞介导的肿瘤免疫抑制治疗策略[14]。
另一方面, 基于A20在胶质母细胞瘤干细胞(germline stem cells, GSCs)和胶质瘤细胞中高表达的特点, 有研究开始了利用RNA干扰靶向抑制A20表达分析其抗肿瘤作用以及作为靶向治疗的可能性, 体外实验显示A20-siRNA可以抑制GSCs和胶质瘤细胞株增殖, 诱导细胞凋亡, 体内试验显示小鼠肿瘤生长抑制、生存期延长[18, 26]。因此, 针对A20在不同肿瘤中的不同作用机制, 选择性抑制A20则是对于部分A20高表达肿瘤包括未分化鼻咽肿瘤、低分化头颈部鳞状细胞癌、雌激素受体和孕激素受体阴性乳腺癌等的选择性靶向治疗方向。
6 展 望
A20从一个去泛素化作用的抗炎相关因子到被确定为肿瘤抑制因子的过程, 实际上也是近年来逐步认识慢性炎症向肿瘤发生转化相关机制的过程, 对于淋巴肿瘤常常累及A20失活更值得我们进一步探讨A20在免疫细胞中的特定作用及其诱导淋巴肿瘤发生中可能的特殊机制。而在治疗性研究方面, 针对A20功能的双重性, 值得深入考虑其作为靶向分子治疗的应用价值和可能存在的负面效应。根据野生型A20的补充可诱导淋巴瘤细胞重新获得凋亡功能的特点以及A20基因异常改变与CLL预后的相关性, 将其作为一个生物标志物引入临床预后分析和相应的靶向基因治疗策略, 可能是目前值得探索的新方向。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|