{kind=link}
RAS突变在EML4-ALK阳性肺癌联合治疗中的作用
引用本文
评价者:黄诚, 林根, 文献合成者:黎扬斯. RAS突变在EML4-ALK阳性肺癌联合治疗中的作用.循证医学, 2015,16(1): 36-40
Reviewers:HUANG Cheng, LIN Gen, Literature Co-worker: LI Yang-si. RAS Mutation Underlies a Rational Polytherapy Strategy in EML4-ALK-Positive Lung Cancer. Journal of Evidence-based Medicine, 2015,16(1): 36-40
Permissions
Reviewers:HUANG Cheng, LIN Gen, Literature Co-worker: LI Yang-si. RAS Mutation Underlies a Rational Polytherapy Strategy in EML4-ALK-Positive Lung Cancer. Journal of Evidence-based Medicine, 2015,16(1): 36-40
Copyright©2016, 《循证医学》编辑部
《循证医学》杂志 版权所有
RAS突变在EML4-ALK阳性肺癌联合治疗中的作用
 | 黄诚, 福建省肿瘤医院肿瘤内科主任, 主任医师, 福建医科大学肿瘤内科教研室主任, 教授。福建省肿瘤化学治疗质量控制中心主任, 兼任中国抗癌协会肺癌专业委员会常委, 中国癌症康复与姑息治疗专业委员会副主任委员。中国肿瘤临床学会(CSCO)常务理事、小细胞肺癌专家委员会副主任委员、中国肿瘤驱动基因分析联盟常务委员、血管靶向治疗专家委员会委员及生物标志物专家委员会委员。中国药学会抗癌药物专业委员会委员。福建省抗癌协会常务理事, 福建省肺癌专业委员会主任委员, 福建省肿瘤内科专业委员会主任委员, 福建省癌症康复与姑息治疗专业委员会前任主任委员; 《中国肺癌杂志》、《临床肿瘤学杂志》、《中国肿瘤临床与康复》、《JTO》中文版等杂志编委。长期从事肿瘤内科的临床和科研工作, 并且参加30多项国际多中心临床研究项目, 主持省部级课题3项, 在省级以上核心刊物发表学术论文70多篇, 获省级科技成果奖2项。 |
关键词:
肺癌; RAS突变; EML4-ALK融合阳性; 治疗
中图分类号:R734.2
文献标识码:A
收稿日期:2016-01-14
RAS Mutation Underlies a Rational Polytherapy Strategy in EML4-ALK-Positive Lung Cancer
Key words:
lung cancer; RAS mutation; EML4-ALK fusion-positive; therapy
1 文献来源
研究一:Westcott PMK, Halliwill KD, To MD, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer[J].
Nature, 2014, 517(7535): 489-492.
研究二:Hrustanovic G, Olivas V, Pazarentzos E, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer[J]. Nat Med, 2015, 21(9): 1038-1047.
2 证据水平
研究一:1b; 研究二:2a。
3 背 景
• 研究一:驱动基因的陆续发现, 提示人类肿瘤的发生与环境暴露有关。
• 研究二:体外实验表明RAS-MAPK通道的活化与ALK抑制剂耐药相关; 合理的联合靶向治疗可能对EML4-ALK融合阳性肺癌获得性耐药有抵抗作用。
4 目 的
• 研究一:探讨致癌物与KRAS突变选择的作用, 环境暴露与肿瘤发生机理之间关系。
• 研究二:探讨RAS-MAPK通道活化在ALK抑制剂耐药的作用、ALK与MEK抑制剂联合对ALK融合肺癌患者的作用。
5 研究设计
研究一:
• 研究条件:加州大学旧金山分校动物实验研究中心。
• 研究方法:对小鼠模型肿瘤进行基因提取, 分别行全外显子测序, 单核苷酸变异(single-nucleotide variations, SNVs)的鉴别及确认、突变谱分析、识别潜在的驱动基因、FREEC测序数据库评估拷贝数、小鼠与人肿瘤突变的对比。细胞学:行Mtus1基因敲除及MTT的评价, 并结合TCGA数据库进行生存分析。
• 研究对象:三种非小细胞肺癌(non-small cell lung cancer, NSCLC)FVB/N小鼠模型。致癌物模型组:甲基亚硝基脲模型(MNU)(44个肿瘤)及尿烷(urethane)模型(26个肿瘤); 基因工程诱导模型:KRASLA2模型(12个肿瘤)。并用细胞学进一步验证。
• 干预措施:将三种模型肿瘤进行全外显子测序等分析。
• 评价指标:对比致癌物与基因工程诱导的肿瘤发生原理, 包括异倍体、拷贝数及SNVs、原始驱动基因、抑癌基因等, KRAS基因突变的选择性。
研究二:
• 研究条件:加州大学旧金山分校医学中心及Helen Diller 癌症中心。
• 研究方法:对ALK-TKI(克唑替尼、色瑞替尼)耐药肺腺癌细胞株进行外显子测序, 体外耐药评估、免疫印迹、siRNA分析、病毒转染、shRNA分析、DNA转染等分析; 构建移植瘤小鼠模型, 评估ALK与MEK抑制剂联合治疗的疗效。
• 研究对象:H3122、STE-1、H2228肺腺癌细胞株, 并构建克唑替尼耐药细胞株、色瑞替尼耐药细胞株; NOD/SCID小鼠。
• 干预措施:小鼠模型共分三组。色瑞替尼组:连续5天应用25 mg/kg或50 mg/kg 色瑞替尼; 曲美替尼组:连续5天应用1 mg/kg或3 mg/kg曲美替尼; 联合用药组:连续5天应用色瑞替尼25 mg/kg或 50 mg/kg以及曲美替尼1 mg/kg。
• 评价指标:EML4-ALK与MAPK通路的关系、RAS-MAPK通路活化的途径、ALK + MEK抑制剂联合治疗的疗效。
6 主要结果
研究一:
• 致癌物与基因工程诱导的肿瘤发生原理可能不同:与致癌物模型比较, KRASLA2小鼠表达更多的异倍体、拷贝数及SNVs(见图1)。
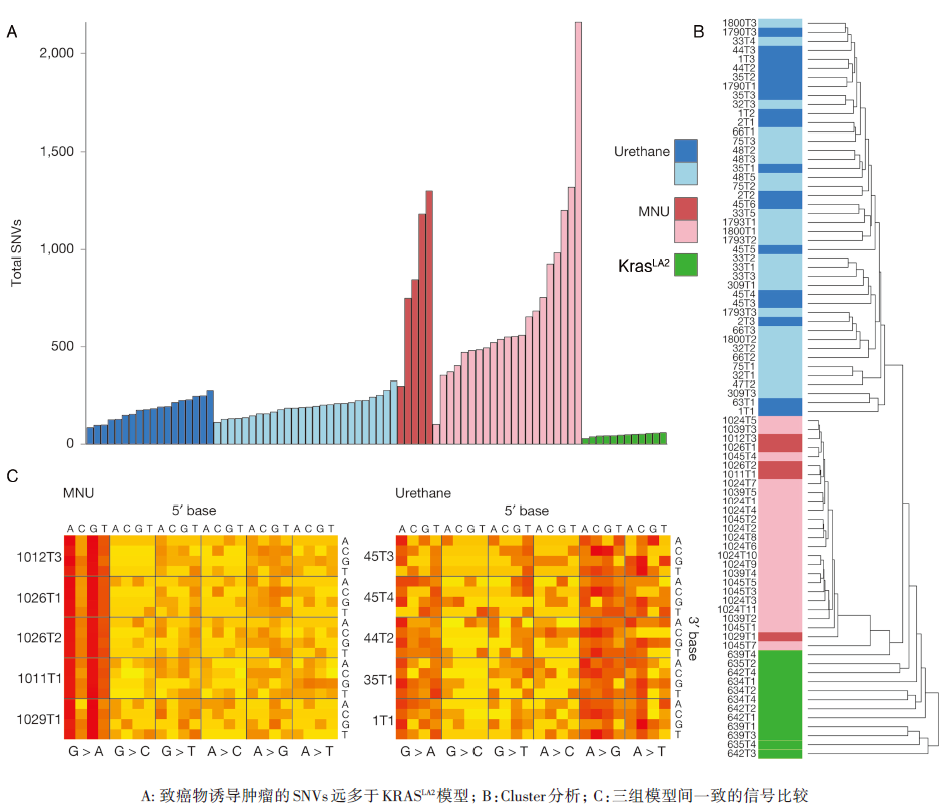 | 图1 致癌物模型与驱动基因模型的突变负荷和突变谱的比较 |
• 肿瘤分化过程中, KRAS基因突变存在选择性:尿烷小鼠野生型模型94% 为KRAS Q61R突变, 而KRAS杂合小鼠模型则92%为 KRAS Q61L突变。
• 致癌物诱导模型中, 外显子突变图谱提示广泛的原始驱动基因信号; 腺癌则在CpG位点新增C> T突变。
• 敲除抑癌基因Mtus1可使得受KRAS G12D驱动的肺癌细胞株生长加速, 而TCGA数据库分析Mtus1高表达肺癌患者生存期延长。
研究二:
• MAPK可通过KRASWT 拷贝数增加或MAPK磷酸化DSUP6表达减少再活化, 而这两个途径都与EML4-ALK抑制剂在肺癌中获得性耐药相关。
• 在ALK阳性肺癌模型上, 预防性联合应用ALK抑制剂和MEK抑制剂可提高药物疗效。
7 结论
研究一:致癌物与基因工程诱导的KRAS突变模型在肿瘤发生机理存在差异; 小鼠肿瘤模型对研究人肿瘤发生机制具有重要作用。
研究二:RAS-MAPK信号通路在EML4-ALK融合肺癌获得性耐药中起着关键作用; 联合ALK及MEK抑制剂可有效延缓耐药发生。
8 评论
Balmain及其团队应用二代测序技术对三种不同方法诱导的KRAS突变NSCLC的小鼠模型进行全外显子组测序, 并与TCGA数据库中的人肺腺癌全外显子组测序结果进行比较, 研究结果对我们今后的基础和临床研究可能主要有以下几点指导意义。
第一, 研究采用的三种小鼠模型, 一种是基因工程诱导的KRAS突变(G12D)肺癌小鼠模型, 另外两种是通过致癌物甲基亚硝基脲或尿烷诱导的KRAS突变肺癌小鼠模型。全外显子组测序结果显示基因工程诱导的小鼠模型与致癌物诱导的小鼠模型的癌基因组学图谱存在着明显差别:致癌物诱导的小鼠模型的单核苷酸变异的数目要远高于基因工程小鼠模型, 呈10倍到上百倍的增高, 相反, 基因工程诱导的小鼠模型的基因组非整倍体以及基因拷贝数改变要显著高于致癌物诱导的小鼠模型。这些数据充分说明了基因工程诱导与致癌物诱导的小鼠模型可能通过不同的途径诱导肿瘤的发生发展, 这一结果对今后在药物研发的临床前研究过程中如何选用恰当的动物模型具有一定的警示作用。
近年来, 肿瘤治疗手段层出不穷, 尤其是靶向治疗药物的研发呈井喷态势。尽管如此, 仍然只有很少比例的研发药物获得批准上市, 这其中也包括了一些动物模型中显示出良好临床应用前景的药物, 这一现象提示临床前研究所采用的动物模型可能在预测临床实际疗效方面仍然存在局限性。在临床实践中, 具有相同驱动基因突变的患者对同一种靶向药物的治疗疗效存在明显的异质性, 癌基因组的背景差异可能是导致这种现象的主要原因之一。举例而言, 一项多中心、随机、安慰剂对照的Ⅱ 期临床试验显示MEK抑制剂司美替尼联合多西紫杉醇二线治疗KRAS突变NSCLC具有潜在的临床应用前景[1], 相关的一项基础研究结果提示这一联合治疗模式的疗效与抑癌基因P53与LKB1的状态密切相关, KRAS突变合并LKB1缺失的动物模型对这种联合治疗模式高度抵抗[2]。因此, 选用适合的动物模型进行临床前研究可能对预测临床实际疗效以及后续的临床研究产生较大影响。
研究者将致癌物诱导的KRAS突变肺癌小鼠模型的基因图谱与TCGA数据库中的人肺腺癌全外显子组测序结果相比较, 发现驱动基因突变谱在两组数据库中有相当一部分存在重叠现象, 这些研究结果提示致癌物诱导的KRAS突变肺癌小鼠模型似乎更符合人腺癌临床前研究的模型要求。近期, Day等在《Cell》杂志上发表了一篇关于临床前研究模型的综述, 值得我们进一步学习借鉴[3]。
第二, 这个研究还有一个令人感兴趣的现象是:在致癌物诱导的小鼠模型中, 不同致癌物诱导的KRAS突变类型不一样, 甲尿烷诱导的KRAS突变基本都是G12D, 而尿烷诱导的KRAS突变绝大多数是Q61表型; 另外, 同样是尿烷诱导的KRAS突变模型, 胚系KRAS基因状态决定了肿瘤KRAS基因是发生Q61R还是Q61L突变(类型)。这些研究结果提示我们:不同的KRAS突变类型可能并不是一个随机选择的结果, 而是外界环境暴露与自身基因状态共同作用决定的。目前, 我们对同一个驱动基因不同突变类型的认识正在不断加深, 但目前大部分局限在不同突变类型的生物学行为[4, 5]以及治疗疗效的差别[6, 7]等角度。正如该文献作者所提出的, 不同突变类型的动物模型可以为我们提供研究环境暴露与肿瘤发生发展的演变过程的良好研究模型, 另外, 在此基础上, 在全基因组背景下通过对同一基因不同突变类型的发生机制的深入认识, 有助于向更高层次的精准治疗迈进。
ALK融合基因是肺腺癌常见的驱动基因之一, 其中EML4-ALK这一亚型占绝大多数。Bivona及其研究团队关注于EML4-ALK这一常见亚型, 在大量细致的研究工作基础上得出以下几点关键结论, 对我们今后的临床实践均有一定的指导意义。
第一, 该研究发现EML4上的HELP结构域可能是EML4-ALK融合蛋白在细胞膜锚定以及特异性激活RAS途径所必需的。以往对ALK融合基因的结构与功能研究主要局限在癌基因ALK上, 对EML4融合片段的生物学功能的了解并不透彻。完整的ALK蛋白具有跨膜结构域, 对细胞膜上的锚定起到关键性作用, 但EML4-ALK融合基因缺乏ALK跨膜结构域, 该研究首先应用免疫荧光染色证实内源性EML4-ALK可以锚定于细胞膜上, 为了探明EML4-ALK中何种结构在细胞膜锚定上起关键作用, 研究者进一步构建野生型EML4-ALK过表达细胞模型、EML4-ALK突变模型(缺乏EML4的HELP结构域), 观察上述干预对EML4-ALK细胞膜锚定以及下游RAS途径活化的影响, 通过上述正反两方面的研究, 结果提示EML4上的HELP结构域可能是EML4-ALK细胞膜锚定以及特异性激活RAS途径所必需的。这一现象提示我们:在融合基因的研究中, 我们的关注点不应该仅仅放在癌基因片段上, 不同类型的ALK融合基因其生物学作用是否有所区别, 可能也是今后的一个研究方向。但同时我们应该注意的是, 上述HELP结构域的生物学功能的验证缺乏直接证据, 氨基酸序列的变化可能会改变蛋白质的空间构象, 从而影响真正起关键作用的结构域, 因此, 该研究如果能够增加其他非HELP结构域的突变模型或采用多种蛋白结构功能研究方法进一步验证, 研究结论可能更为可靠。
第二, ALK阳性NSCLC高度依赖RAS-MAPK途径。以往研究表明ALK可以激活多个下游信号途径, 如RAS-MAPK、PI3K-AKT以及JAK-STAT等, 但哪一条途径扮演了关键角色仍未完全明确。在该研究中, 研究者应用MEK抑制剂观察到细胞增殖受到明显抑制, 令人惊讶的是, 这种抑制效应与ALK抑制剂相当, 相反, PI3K-AKT或JAK-STAT途径抑制剂均未观察到抑制细胞增殖效应。
第三, 发现了两种新的ALK抑制剂继发性耐药机制。以往的基础及临床研究已经确定了大部分的ALK抑制剂继发性耐药机制, 这其中包括ALK激酶结构域的点突变、ALK融合基因拷贝数增加以及旁路途径活化等等[8]。该研究发现的耐药机制, 一个是KRAS基因拷贝数增加, 另一个是MAPK通路中负性调节分子双特异性蛋白磷酸酶(DSUP 6)的表达下调, 这两个机制均是通过重新活化RAS-MAPK通路从而产生继发性耐药。上述研究结果在小样本临床病例中也得到了初步证据, 但仍需进一步扩大样本量进行重复临床验证。另外, 5例上述继发性耐药机制的患者接受二代ALK抑制剂色瑞替尼治疗, 4例患者短期内快速进展, 提示目前现有的治疗药物对逆转上述耐药机制的疗效有限, 需要探索该耐药机制的有效治疗策略。
第四, 建立ALK抑制剂和MEK抑制剂联合阻断的治疗策略; 上述研究结果提示无论是初治还是继发性耐药患者, ALK阳性NSCLC均高度依赖RAS-MAPK途径, 另外, 单纯给予ALK抑制剂并不能完全抑制下游信号分子ERK活化, 基于以上原因, ALK抑制剂和MEK抑制剂联合阻断的治疗策略有可能增加疗效。研究者在体外及动物模型中初步验证了这种联合治疗模式可以增强抗肿瘤效应并且延缓耐药发生。
这种联合治疗模式加用低剂量MEK抑制剂, 在提高疗效的同时并不增加明显的毒副反应。目前ALK抑制剂和MEK抑制剂均已有上市药物, 应该尽快进行转化性研究的临床验证步伐。同时, 我们还应看到, ALK抑制剂和MEK抑制剂联合阻断的理论基础之一是单纯ALK抑制剂并不能完全抑制ERK活化, 但这一现象是否仅仅是由于ALK抑制剂本身作用活性偏低的问题, 能否通过研发作用活性更强的新一代ALK抑制剂就可以获得比联合治疗模式更好的临床疗效?在这一方面, 二代EGFR-TKI阿法替尼和三代EGFR-TKI的代表药物AZD9291针对T790M耐药的研发过程已经为我们提供了类似的经验教训。
实际上, 以往已经有研究表明, ALK抑制剂联合MEK抑制剂有可能提高ALK阳性NSCLC的治疗疗效[9, 10] , 但该研究可能是第一个在充分阐明相关机制的基础上进行联合治疗模式的探索。目前, 通过联合用药进一步增加靶向治疗的疗效已经成为研究热点, 如EGFR-TKI联合化疗、抗肿瘤血管生成治疗等等。但这种联合治疗模式是否成功, 相关基础研究的支持很关键, 而不是盲目地去尝试, 即便是联合治疗模式, 我们也需要个体化精准治疗, Bivona及其研究团队的工作给我们做出了表率。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|