



{kind=link}
{kind=link}
{kind=link}
肿瘤二代测序临床报告解读共识
引用本文
二代测序临床报告解读肿瘤学专家组. 肿瘤二代测序临床报告解读共识. 循证医学, 2022,22(2): 65-79.
The Oncology Expert Group on the Interpretation of Next Generation Sequencing Clinical Reports . Consensus on Interpretation of Clinical Reports for Next-Generation Sequencing of Tumors. Journal of Evidence-Based Medicine,2022,22(2): 65-79.
DOI:10.12019/j.issn.1671-5144.2022.02.001
Permissions
The Oncology Expert Group on the Interpretation of Next Generation Sequencing Clinical Reports . Consensus on Interpretation of Clinical Reports for Next-Generation Sequencing of Tumors. Journal of Evidence-Based Medicine,2022,22(2): 65-79.
DOI:10.12019/j.issn.1671-5144.2022.02.001
Copyright©2022, 《循证医学》编辑部
《循证医学》杂志 版权所有
肿瘤二代测序临床报告解读共识
通讯作者: 张绪超, Tel:020-83827812, E-mail:zhxuchao3000@126.com。
摘要
随着新型治疗药物的研发以及多学科综合治疗模式的优化,传统的病理分型及检测方法已经不足以满足临床需求,二代测序(next generation sequencing,NGS)已成为中国肿瘤医生常用的检测手段。为进一步协助临床医生理解临床靶点或驱动基因相关变异注释及解读、梳理NGS报告的逻辑、提升抓取关键信息,二代测序临床报告解读肿瘤学专家组对国内外NGS检测最新进展进行认真分析、讨论和总结,在《二代测序临床报告解读指引》基础上增加部分NGS报告解读示例,同源重组缺陷(homologous recombination deficiency,HRD)及微小/分子/可测量残留病灶(minimal/molecular/measurable residual disease,MRD)相关内容解读,制定了《肿瘤二代测序临床报告解读共识》,最终帮助临床医生做出正确的临床决策。
关键词:
二代测序; 报告解读; 共识
中图分类号:R446.7
文献标识码:A
Consensus on Interpretation of Clinical Reports for Next-Generation Sequencing of Tumors
Abstract
With the development of new therapeutic drugs and the optimization of multidisciplinary comprehensive treatment models, traditional pathological typing and detection methods are no longer sufficient to meet clinical needs. Next generation sequencing (NGS) has become a commonly used detection method by Chinese oncologists. In order to further assist clinicians to understand the annotation and interpretation of variants related to clinical targets or driver genes, sort out the logic of NGS reports, and improve the capture of key information, The Oncology Expert Group on the Interpretation of NGS Clinical Reports performed careful analysis, discussion, and summary based on the latest progress in NGS testing at home and abroad, and added some examples of NGS report interpretation, as well as interpretation of homologous recombination deficiency (HRD) and minimal/molecular/measurable residual disease (MRD) related content according to the “Guidelines for the Interpretation of Next-Generation Sequencing Clinical Reports”, and formulated the “Consensus on Interpretation of Clinical Reports for Next-Generation Sequencing of Tumors” to help clinicians make the right clinical decisions.
Key words:
next generation sequencing; report interpretation; consensus
背景
随着肿瘤精准治疗时代的到来, 常规的病理学诊断方法已无法满足临床需求, 国内外多项指南和共识均认可基于二代测序(next generation sequencing, NGS)全面分子检测对于指导临床治疗的价值, 全面而准确的肿瘤基因诊断结果已成为临床诊疗的刚需。基于此, 为进一步提升临床医生NGS报告解读能力, 二代测序临床报告解读专家组在学习全球最新科研成果的基础上, 参考国际指南和中国国情, 经多学科专家组的反复讨论撰写了《二代测序临床报告解读指引》。此次二代测序临床报告解读肿瘤学专家组回顾临床病例, 挑选了其中部分典型案例进行解读。同时也对国内外NGS检测最新进展进行认真分析、讨论和总结, 增加了同源重组修复缺陷(homologous recombination deficiency, HRD)及实体瘤微小/分子/可测量残留病灶(minimal / molecular / measurable residual disease, MRD)相关内容, 制定了《肿瘤二代测序临床报告解读共识》, 旨在帮助临床医生梳理NGS报告解读逻辑, 快速抓取关键信息, 同时尽可能规避不正确的解读基因组信息导致的潜在危害, 最终做出正确的临床决策, 为患者带来更多的临床获益。
正文
每个肿瘤基因组中可能存在数百至数千个体细胞突变, 其中许多是相对个体化的变异。对NGS鉴定出的多个基因变异(genomic alterations, GA)进行优先级排序是一项重大挑战。这些基因变异主要包括单核苷酸变异(single-nucleotide variant, SNV/single-nucleotide polymorphism, SNP)、短片段插入/缺失突变(Indel)、重排(融合)、拷贝数变异(copy number variation, CNV)及其他复杂突变等[1]。部分变异出现在生物学及临床相关、甚至是分子治疗潜在靶标的肿瘤基因中, 但并非所有肿瘤相关基因发生的变异均为(潜在)功能性变异, 更多的基因变异尚无明确的生物学和/或临床意义。随着高通量测序分析进入临床领域, 产生了大量数据, 而如何及时、准确的将测序发现的肿瘤基因组变异信息转化为临床医生可读取并用于指导临床决策的结构化循证报告(structured evidence-based reports), 正变得越来越重要[2]。本次从临床靶点或驱动基因相关体细胞变异注释及解读、NGS报告解读及临床决策、可报告范围及质量控制3个方面对NGS临床报告解读作以指引, 并增加HRD及实体瘤MRD相关解读内容, 以更加贴合国内外最新进展。
1 临床靶点或驱动基因相关体细胞变异注释及解读
基于NGS技术检测肿瘤体细胞变异的实验流程可概括为以下几个主要环节:样本采集及质量控制、DNA提取、文库制备、测序及基因组数据生成。而数据分析可进一步拆解为3个流程:变异识别(variant identification)、变异注释及过滤(variant annotation and prioritization)、变异的临床解读(interpretation of clinical significance)。其中, 变异识别、注释及过滤经由生物信息学工具实现; 而临床解读则需要基于严格的分级逻辑, 整合当前公共数据库及已发表文献的海量信息, 特别是变异-药物敏感性信息, 建立基因变异的临床解读知识库, 最终将与送检样本的对应癌种及检出的基因变异相匹配的临床意义(如药物敏感性信息)及其证据级别呈现在NGS报告中[3, 4, 5]。
目前有多个循证分级系统可用于指导基因体细胞变异的临床解读, 包括2017年美国分子病理学协会(Association for Molecular Pathology, AMP)/美国临床肿瘤学会(American Society of Clinical Oncology, ASCO)/美国病理学家协会(College of American Pathologists, CAP)联合制定的体细胞变异解读指南; 2018年欧洲肿瘤内科学会(European Society for Medical Oncology, ESMO)发布的分子靶点临床可操作性量表(ESMO Scale for Clinical Actionability of molecular Targets, ESCAT); 以及纪念斯隆-凯特琳癌症中心(Memorial Sloan Kettering Cancer Center, MSKCC)的精准医疗肿瘤数据库(Precision Oncology Knowledge Base, OncoKB)证据等级规则。总体而言, 无论哪个分级系统都遵循一些共性原则, 包括循证、跨癌种处理等, 故其中并无优先推荐者。临床医生在阅读1份NGS报告时应先了解其变异解读依据的证据分级原则及其采用知识库的局限性, 以帮助自己更好的理解报告内容。
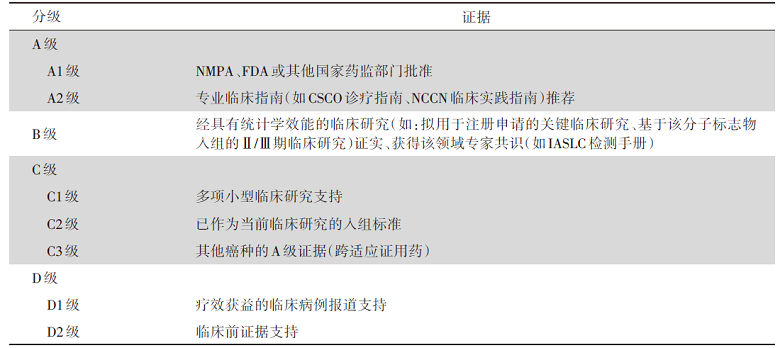
根据2017年AMP/ASCO/CAP联合制定的体细胞变异解读指南[6], 体细胞变异在不同癌种中对应的药物敏感性证据分为4个等级:A级, 美国食品药品监督管理局(Food and Drug Administration, FDA)批准或专业临床指南推荐; B级, 经具有足够统计学效能的临床研究证实、获得该领域专家共识; C级, 其他癌种中的A级证据(跨适应证用药)、或已作为临床试验的入组标准; D级, 临床病例报道或临床前证据支持。体细胞变异对特定肿瘤的诊断及预后价值, 亦给出相应分级:A级, 专业指南中定义的特定肿瘤的诊断/预后因子; B级, 经具有足够统计学效能的临床研究证实其诊断/预后价值; C级, 多项小型研究支持其诊断/预后价值; D级, 小型研究或个案报道提示其辅助诊断/预后价值(独立或联合其他标志物)。
基因变异按照其临床意义的重要性分为4类变异:Ⅰ 类变异, 有重要的临床意义, 具有A级或B级证据; Ⅱ 类变异, 有潜在的临床意义, 具有C级或D级证据; Ⅲ 类变异, 临床意义不明; Ⅳ 类变异, 无害或可能无害, 详见图1。
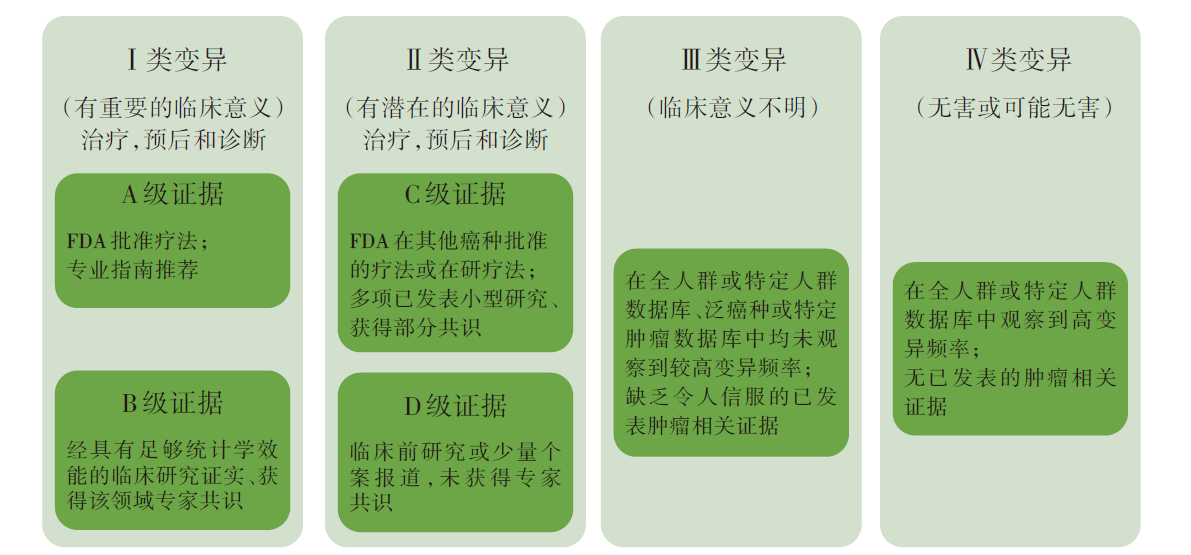 | 图1 AMP/ASCO/CAP指南:基于证据的体细胞突变分类[6]Fig.1 AMP/ASCO/CAP guidelines: Evidence-based variant categorization[6] |
该体细胞变异解读指南在国内影响范围最广, 许多第三方NGS检测公司的临检报告即遵循该指南的分级原则对基因变异进行解读, 详见表1。
| 表1 NGS报告体细胞突变证据分级标准 Tab.1 Categories of clinical and/or experimental evidence for the interpretation and reporting of somatic variants |
ESCAT是由ESMO转化研究和精密医学工作组(Translational Research and Precision Medicine Working Group, TR and PM WG)发起建立的、基于临床靶标相关证据的基因变异临床分类系统[7], 于2018年发表, 将基因变异分为6个级别:Ⅰ 级, 可用于常规临床决策的靶点, 如乳腺癌的HER2扩增和非小细胞肺癌(non-small cell lung cancer, NSCLC)的EGFR敏感突变, 其中Ⅰ -A级突变是基于前瞻性临床研究已有了显著生存获益的靶标突变类型; Ⅱ 级, 已有证据表明患者将受益于针对性靶向治疗, 但仍需更多数据证实, 如PI3K通路中的AKT1、PTEN突变; Ⅲ 级, 在其他肿瘤类型(而非该患者肿瘤类型)中已证实临床获益的靶标性基因变异, 如NSCLC以外肿瘤中的EGFR19del突变; Ⅳ 级, 仅有临床前证据支持靶标性的基因变异; Ⅴ 级, 有证据表明针对此类变异的靶向治疗可获得客观缓解, 但缺乏有临床意义的生存[无进展生存期(progression-free survival, PFS)或总生存期(overall survival, OS)]获益, 或可支持联合治疗策略; Ⅹ 级:已证实缺乏临床价值(而非“ 尚无证据支持” ), 不应影响临床决策。
OncoKB是由MSK癌症中心维护的精准医疗肿瘤数据库。该数据库旨在提供详细的、基于证据的基因组变异相关信息, 以帮助临床医生进行治疗决策。数据基于FDA、NCCN、ASCO指南等权威资料及其他科学文献、以及疾病专家小组的建议, 对不同变异的药物预测价值进行证据分类[8]:1/2级, FDA认可、或被认为是临床标准(standard of care, SOC)生物标志物, 可预测在特定疾病背景下对已获批药物反应的变异; 3级, 基于对临床试验中待测目标药物的有希望的临床数据, 被认为可预测药物反应的变异; 4级, 根据对临床试验中待测目标药物的令人信服的生物学证据, 被认为可预测药物反应的变异。
2 NGS报告解读及临床决策
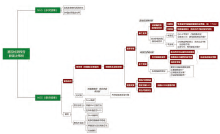
一份结构化循证报告的背后, 需要基于特定逻辑建立临床解读知识库框架, 并通过对开源知识库及海量科学文献的信息甄别、分级、编辑, 不断完善机构内部临床解读知识库。通过生物信息分析流程将基因变异识别、注释、过滤后, 可报告变异进入临床解读知识库进行变异、癌种、证据匹配, 形成可读的结构化NGS临床报告[9], 最终对其进行解读及实施临床决策, NGS报告解读决策树详见图2。
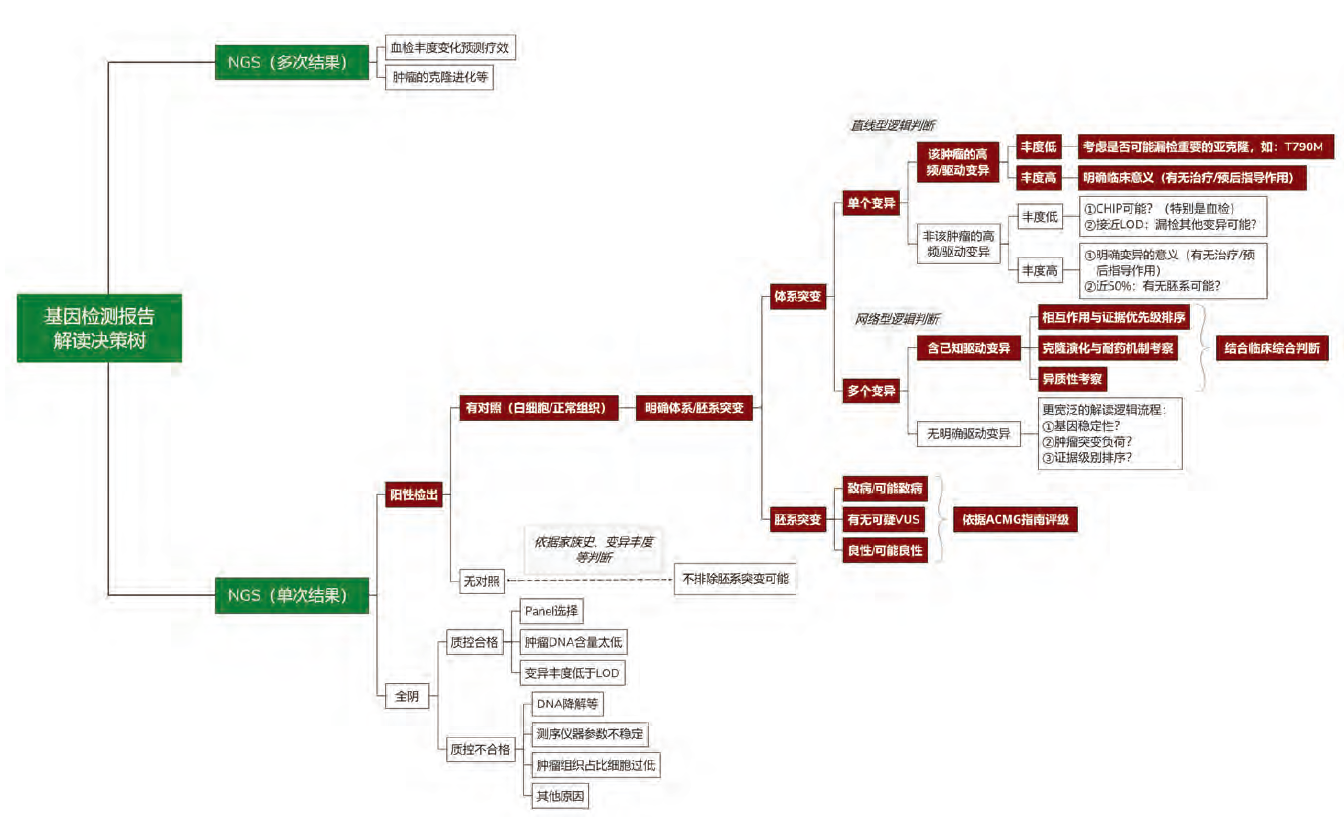 | 图2 NGS报告解读决策树 |
2.1 针对“ 全阴报告” 的解读逻辑
当临床医生面对1份体细胞变异“ 全阴报告” (即1份样本未能检出任何肿瘤体细胞突变)时, 应首先考虑以下两点:
(1)送检样本是肿瘤组织、外周血循环游离DNA(circulating free DNA, cfDNA)或其他; 结合样本质控信息及患者治疗史, 综合判断样本中是否含有足够肿瘤成分, DNA总提取量和/或肿瘤占比是否可能低于NGS的LOD。
(2)选择的NGS panel(尤其是仅包含数个至数十个基因热点区域的小panel)是否与患者的肿瘤类型相匹配, 即该肿瘤常见变异是否能被该panel覆盖。
以上评估可以帮助我们判断, 该“ 全阴” 结果是提示“ X基因野生型” 可能性大, 还是“ X基因状态未知” 可能性大。比如1份血检全阴报告, 采用panel针对泛癌设计、覆盖上百基因的热点区域, 对目标癌种常见变异的覆盖度> 90%, 此时的全阴, 很可能仅提示肿瘤全身负荷较低或其他原因(如患者正接受有效的抗肿瘤治疗)导致释放入血的循环肿瘤DNA(circulating tumor DNA, ctDNA)含量极低、未达检测平台的LOD。这种“ 全阴” 不能反映肿瘤基因组变异状态, 仅能提示“ 基因突变状态未知” 。当然, 这种血检全阴报告可以提供突变谱以外的其他信息, 在特定场景下提示一定的预测、预后价值。
2.2 针对NGS报告中基因融合、外显子跳读突变检测的说明与突变丰度及拷贝数的解读逻辑
基因融合在血液肿瘤或者实体瘤中均可检出, 目前FDA及NMPA针对ALK、ROS1、RET、FGFR2和NTRK1/2/3等融合靶点已经批准了多款相应的靶向药物。由于DNA测序(DNA sequencing, DNA-seq)检测融合需要同时覆盖外显子和内含子, 而内含子区域常见重复区、同源区, 干扰DNA-seq检测灵敏度; 同时有些基因断点可能发生在多个内含子, 且内含子长度大, panel设计往往难以全部覆盖。因此从融合产物功能性预测及检测全面性而言, RNA测序(RNA sequencing, RNA-seq)更灵敏更直接, 同时验证功能。RNA-seq更具优势, 有利于临床应用。
MET14外显子跳读突变是已明确的NSCLC驱动变异, 约3%~5%的NSCLC携带MET 14外显子跳读突变[10, 11, 12]。日本已批准tepotinib治疗MET 14外显子跳读的NSCLC患者。FDA已批准capmatinib用于MET 14外显子跳读的NSCLC患者。赛沃替尼已获NMPA批准用于携带MET 14外显子跳读的晚期NSCLC患者。由于MET 14外显子发生的突变大部分位于外显子起始端或末端和内含子交接处, 突变分布的区域较广泛, 而以DNA为基础的NGS探针所需覆盖的区域广泛, 一旦不能全面覆盖这些突变的区域, 可能会造成漏检。而在RNA层面上, MET 14外显子发生的突变会直接导致整个14号外显子的缺失, 因此当检测到MET 13外显子后连接的是15号外显子, 就能说明存在14号外显子跳读突变, 探针覆盖区域小, 不容易产生漏检。
MSK癌症中心的1项大样本肺腺癌研究中, 使用DNA进行NGS测序(MSK-IMPACT 468 genes DNA panel), 检出的驱动基因阳性率为76.6%(1 933/2 522), 其中融合阳性率为7.7%(195/2 522), MET 14外显子跳读突变阳性率4.7%(119/2 522)。在DNA-seq检测为驱动基因变异阴性的232例患者中, 经RNA-seq(MSKCC-Fusion)又检测到了36例融合及MET 14外显子跳读突变, 其中融合阳性患者为30例, 6例MET 14外显子跳读突变, 其中10例患者接受了相应靶向治疗, 临床获益率80%[13]。2020年ASCO上公布了1项研究, 比较了DNA检测与RNA检测对MET 14外显子跳读突变的检出率。研究发现, 在对644例肺腺癌患者使用以DNA为基础的NGS检测时, 共检测到16例MET 14外显子跳读突变(2.5%), 而当进一步使用以RNA为基础的NGS技术对剩下的DNA检测阴性样本进一步检测, 又检出了额外9例MET 14外显子跳读突变[14]。
基于以上情况, NCCN的NSCLC指南(2021 v1版)建议, 如果使用DNA检测NGS技术没有检测到任何驱动突变时, 建议补充以RNA为基础的NGS检测, 以增加基因融合及MET 14外显子跳读突变的检出率。未来以DNA与RNA检测相结合为代表的多组学检测, 将会为受检者提供更全面、精准的检测结果, 有望成为基因检测的新方向。
临床医生在拿到1份NGS阳性报告时, 可能对其中检出的突变/重排的丰度(allele frequency/fraction, AF)及CNV的拷贝数(copy number, CN)存在疑问。基于不同的样本类型, 对AF及CN的理解可能存在一定差异。
2.2.1 基于组织样本的NGS检测
首先对AF绝对数值的解读。对于组织检测结果, 可以基于如下公式, 根据X基因突变(或重排)的AF以及样本中的肿瘤占比, 粗略判断该变异在肿瘤中的克隆占比:
X基因变异AF = 肿瘤占比 × 肿瘤中X变异克隆占比 × 0.5(杂合突变)[或1.0(纯合突变)/中性拷贝数LOH]
一般情况下, 肿瘤细胞中的突变, 尤其是癌基因突变(如EGFR、KRAS活化突变, ALK、ROS1重排)以杂合突变存在; 但上述公式未考虑潜在的拷贝数变异(比如EGFR 19del伴扩增)的影响。而抑癌基因的功能缺失性(loss of function, LOF)杂合突变, 则可能由于杂合性缺失(loss of heterozygosity, LOH)的存在而影响克隆占比的估算。拷贝数为2的LOH(中性拷贝数LOH)可类比纯合突变进行肿瘤克隆占比的估算, 而拷贝数缺失的LOH则更加复杂。例如, 1份晚期肠癌组织样本中, 检出NTRK1重排(不伴CNV), AF为10%; 而病理评估提示该样本肿瘤占比约为50%; 根据公式计算(考虑为杂合突变), 携带NTRK1重排的肿瘤细胞仅占所有肿瘤细胞约40%, 提示NTRK1重排仅存在于肿瘤的1个亚克隆。当然, 用于病理评估的样本仅为1张切片, 不能完全代表检测样本整体; 且病理评估还可能受局部肿瘤坏死等因素干扰, 故病理评估结果仅供参考, 由此推算出来的变异克隆占比与实际情况可能存在一定偏差。而当样本无法进行病理评估时(比如病理蜡卷标本), 则只能以该样本中检出变异的最高突变丰度(maxAF)来反推可能的肿瘤占比。比如1份肠癌样本中同时检出KRASG12D突变、SMAD4突变和NTRK1重排, KRAS突变作为maxAF丰度为25%, 反推该样本肿瘤占比约为50%, 以此为基础评估NTRK1重排的克隆性。
其次对AF相对数值的解读。组织样本中不同变异的相对丰度同样可以提供很多信息。比如1份肺癌组织样本中, 同时检出EGFR 19del和ALK重排, 二者相对丰度为5:1; 结合患者既往EGFR-酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI)长期治疗史, 我们有较大把握推断, 该ALK重排为获得性耐药机制, 而非EGFR/ALK双原发肿瘤。另外, 大样本研究显示, 晚期肠癌患者cfDNA样本中检出的可靶向TRK重排(融合)多以亚克隆形式存在, 即对比主干突变maxAF的相对丰度低于50%。
最后对CNV的解读。CNV及基因组上部分区域的扩增或缺失。面对1份NGS报告, 首先需要注意CNV有不同的报告形式, 最常见的包括CN及扩增倍数(ratio)。前者即1份样本提取的DNA混合物(包含肿瘤DNA及非肿瘤来源DNA)中检出的某基因的平均拷贝数(正常CN为2); 后者即前者与2的比值。基于NGS检测得到的CNV的非校正CN或ratio值与荧光原位杂交(fluorescence in situ hybridization, FISH)检测得到的CN值或ratio值之间存在较大差异, 详见表2。
| 表2 CNV不同报告形式:NGS vs. FISH Tab.2 Different reporting forms of NGS and FISH |
在已知肿瘤细胞占比的情况下, 可通过以下公式粗略换算目标基因的肿瘤细胞CNV(即校正CNV):
报告CNV = 校正CNV × 肿瘤占比 + 2 × (1 - 肿瘤占比)
假设肿瘤细胞中目标基因CN为20, 当肿瘤占比仅为10%时, 实际检出CN值仅约为3.8。由于正常DNA的稀释作用, 当1份样本中肿瘤占比低于20%(即maxAF< 10%)时, CNV的检出敏感性显著降低。
2.2.2 基于外周血cfDNA的NGS检测
NGS血检, 检测对象是血浆中的cfDNA。虽然科研领域人们可以通过一些方法(比如根据cfDNA片段长度的差异)评估ctDNA的实际含量, 但在实际临床应用及大多数科研发表中, 仍以血浆样本中检出体细胞变异的maxAF作为ctDNA含量的替代指标[15]。可针对某一肿瘤患者进行不同临床节点的血检动态监控, 以maxAF反映外周血ctDNA含量的动态变化, 以此反映全身肿瘤负荷变化、预测治疗效果及生存预后。比如, 对于初诊晚期肿瘤患者, 基线的ctDNA含量高低与全身肿瘤负荷相关; 而治疗开始后(3~8周)ctDNA的早期变化趋势可预测当前治疗的总体效果, ctDNA快速清零预示更长的PFS及OS。这种变化趋势比影像学评估更加快速敏感, 比如影像学评估同为SD的患者, ctDNA早期清零与长PFS显著相关; 免疫治疗假性进展的患者, 治疗初期即表现为ctDNA快速下降甚至清零[16]。ctDNA水平的监测可以辅助评估对靶向治疗应答情况, 获悉疾病进展及耐药机制, 对指导后续靶向治疗有较强的指导意义。
例如, 1例Ⅳ 期肺癌患者NGS组织活检检出EGFR L858R(突变丰度84.3%)和TP53 R273C(突变丰度44.0%)共突变, 接受厄洛替尼一线治疗, 期间行外周血NGS动态监测, 经过治疗后病情好转, 病灶缩小, 胸水明显减少, 临床评价疾病稳定(stable disease, SD)(病灶缩小), 血检EGFR L858R(突变丰度3.21%)和TP53 R273C(突变丰度0.22%), 经厄洛替尼治疗1年后复查血检NGS发现EGFRL858R和TP53 R273C突变丰度明显升高(50.99%, 7.20%), 同时检出较高丰度的EGFR T790M突变(7.60%)。患者2个月后出现咳嗽, 复查影像学提示临床疾病进展, 提示NGS血检基因异常的发现可能比临床进展更早, 为加强随访, 甚至提前调整治疗提供重要参考。随后根据NGS血检结果(EGFRT790M突变)给予二线奥希替尼靶向治疗1个月后, 病灶缩小, 复查NGS血检EGFR T790M、EGFR L858R和TP53 R273C突变丰度均明显下降(0.14%、5.28%和0.61%), 与临床病情变化相符。
另外需要注意的是, 由于多数肿瘤患者外周血maxAF< 10%, 对CNV的检测敏感性明显低于突变、重排等变异形式。
2.2.3 基于外周血ctDNA MRD的检测
MRD具有多种表述:微小残留病灶(minimal residual disease)、可测量残留病灶(measurable residual disease)和分子残留病灶(molecular residual disease)。其概念由血液肿瘤逐步延伸至实体肿瘤, 主要指经过治疗后, 传统影像学或实验室方法不能发现, 但通过液体活检发现的癌来源分子异常, 是体现在治疗后体内存在残留肿瘤细胞的生物标志物。由于残留的癌细胞数量较少, 无法通过传统方法检测到。因此, MRD检测对于检测技术的敏感性有极高的要求。随着分子生物学技术的发展, 特别是NGS技术的深入应用, 对MRD的检测已应用到大量临床研究中。
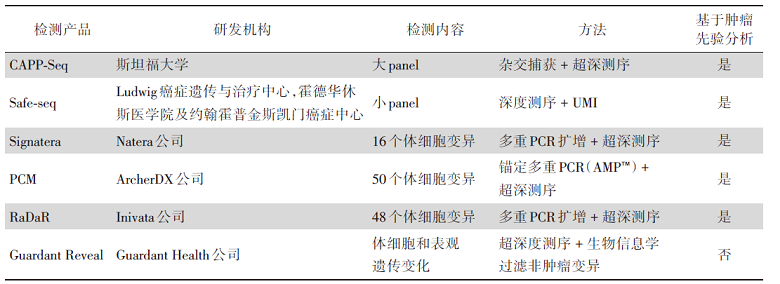
在实体肿瘤中, MRD与患者术后生存复发之间相关性证据已在乳腺癌、消化道肿瘤、肺癌等中得到印证。基于ctDNA的MRD检测已成为临床越发关注的一个重点。很多研究表明MRD能够早于传统影像学发现肿瘤复发, 是一个优良的预后指标, 也有越来越多的证据表明MRD有望成为一个预测指标。目前, 常用的MRD检测技术包括数字聚合酶链式反应(digital polymerase chain reaction, dPCR)、NGS等。其中, NGS作为新兴的MRD检测方法, 对于预后有更准确的评估, 已受到国内外专家认可并推荐。基于NGS检测MRD检测的基本策略, 包括肿瘤先验分析(tumor-informed assays, 个体化定制或NGS panel)和肿瘤未知分析(tumor-agnostic assays, NGS panel和多组学技术), 两种策略目前均处在探索阶段, 需要前瞻性研究确定其敏感性、特异性和临床预测及应用价值。虽然2021年《非小细胞肺癌分子残留病灶专家共识》已提出的标准:基于NGS的突变检测技术, 所选用的多基因panel中必须覆盖患者Ⅰ /Ⅱ 类基因变异, 基本技术标准是可稳定检测出丰度≥ 0.02%的ctDNA[17]。但因目前各家检测机构其技术路线、检测性能、生信分析、MRD阳性标准尚不能统一, 业界也尚未形成标准, 故目前尚无法对MRD检测报告形成统一的解读模式。目前国际上基于NGS开发检测MRD的主要产品见表3[18]。
| 表3 基于NGS检测MRD的主要产品 Tab.3 Representative products of MRD detection based on NGS |
虽然MRD本身仍有很多问题有待进一步解决, 但MRD有望成为未来实体瘤治疗的标志物, 对于早期患者根治性切除术后, 通过MRD的检测及监测予以判断预后, 指导辅助治疗等。而对于晚期患者, 在获得完全缓解或者寡转移经局部治疗后, 合理地通过MRD监测给予药物假期(drug holiday), 理论上可以延缓肿瘤靶向治疗耐药克隆的发展, 延缓耐药, 同时可以减轻患者治疗负担[19]。
2.3 针对单个变异对比多基因多变异的解读逻辑
根据不同NGS panel的可报告范围, 1份NGS阳性报告中的变异可能有多有少。对于不同情况, 临床医生应采取不同的解读逻辑流程。对于多基因变异、特别是多个潜在驱动变异共存的情况, 要结合肿瘤类型、既往治疗史、相对突变丰度、既往分子检测结果等信息综合判读, 以推测不同变异之间的逻辑关系(原发性vs. 获得性, 主克隆vs. 亚克隆, 敏感克隆vs. 耐药克隆), 指导后续治疗。
例如, 1例初诊晚期肺腺癌患者, 行组织NGS检测发现其携带EGFR L858R突变(突变丰度56%), 同时也携带ERBB2基因S310F突变(突变丰度12%), 从NGS检测结果推测携带EGFRL858R突变的细胞为肿瘤主克隆, 而ERBB2仅为致病性相对较弱的亚克隆。该患者接受一线奥希替尼治疗疗效为部分缓解(partial response, PR), PFS达5个月。
另外, 临床上关于判断多原发癌是一直存在的难点, 目前关于多原发癌与转移癌的区分并没有金标准, IASLC提供了一些参考标准:(1)组织学类型:不同的组织学类型可以考虑为是多原发, 比如1个是鳞癌, 1个是腺癌; 但是相同的组织学类型并不能明确是转移癌; (2)遗传特征:用PCR, FISH, NGS等方法检测不同肿瘤的遗传特征, 而基因型完全相同考虑可能是同一个肿瘤发展而来; (3)建议整合所有的信息来进行综合判断, 包括影像学、活检、临床特征、分子特征等, 不同的因素有不同的权重, 应通过多学科会诊来判断[20]。例如, 1例患者查体发现双上肺结节, 胸部CT考虑恶性肿瘤性病变, 完善上腹部增强CT、头颅MR、ECT骨扫描, 均未见明显转移。胸腔镜下行同期双上肺肿瘤肺楔形切除, 系统淋巴结清扫术。虽然术后病理诊断:左上肺浸润性腺癌, 右上肺浸润性腺癌, 腺泡型为主, 淋巴结未见转移。但双侧肿瘤组织分别行NGS检测发现两侧病灶携带不同的肿瘤驱动基因变异(左侧CD74-ROS1融合, 右侧EGFR L858R突变)。经多学科会诊, 参考NGS结果, 最终诊断为双原发肺腺癌。该病例提示多原发癌诊断时须本着谨慎的态度, 结合肿瘤影像学表现、组织亚型、驱动基因变异等进行综合分析, NGS检测能协助提高诊断准确度。
2.4 胚系突变分级注释
当无配对样本(白细胞或正常组织)时, 肿瘤样本(tumor only)的检测在变异注释及过滤过程中, 需要建立生物信息学算法, 以有效区分肿瘤体细胞突变和胚系突变, 确认并过滤胚系多态性。如同时检测配对样本, 则可明确区分体细胞突变和胚系突变。对检出的胚系突变, 应参照ACMG推荐的胚系突变解读流程进行注释及解读[21]。基于当前证据, 胚系变异的致病性分为5级:5级, 致病; 4级, 可能致病; 3级, 意义未明(VUS); 2级, 可能不致病; 1级, 不致病。仅有4/5级胚系变异具有相应的临床指导价值。比如谈及聚腺苷二磷酸核糖聚合酶[poly (ADP-ribose) polymerase, PARP]抑制剂与BRCA1/2胚系突变的相关性, 或BRCA相关卵巢癌-乳腺癌的风险评估, 均默认仅针对4/5类BRCA1/2突变。
2.5 NGS可报告的基因组标签
2.5.1 肿瘤突变负荷
肿瘤突变负荷(tumor mutational burden, TMB)即肿瘤基因组编码区包含的非同义突变的数量或密度(突变数/Mb), 是肿瘤新抗原负荷的替代指标, 过去以全外显子组测序(whole-exome sequencing, WES)作为评估的金标准。目前基于NGS大panel检测的TMB已可达到与WES的高度相关并在大量免疫治疗临床研究中证实其对疗效的预测价值[22]。目前, 业界对组织TMB检测有几点共识:(1)编码区覆盖大于1 Mb(大概相当于300个以上基因的全外显子区域), 最低有效测序深度应≥ 500× 。建议进行TMB检测的靶向测序panel尽可能涵盖患者更多的其他分子遗传信息, 包括可指导靶向治疗的驱动基因突变、免疫治疗可能相关的正向预测因子及负向预测因子; (2)基于靶向测序panel的TMB检测应以WES检测为金标准, 纳入影响蛋白质编码的体细胞突变, 应保证检出突变频率≥ 5%的体细胞突变, 以保证TMB检测值的准确性和稳定性。Panel检测区域可影响TMB值, 应通过至少1 000例WES数据予以校正。同时建议使用对照样本过滤胚系变异; (3)同一份样本采用不同检测平台、不同大小panel、不同的生物信息学算法, 都可能导致TMB绝对数值的差异。因此, 在缺少桥连试验的前提下, 直接采用某一家公司/产品的肿瘤突变负荷高(TMB high, TMB-H)界值(cut-off)来定义其他检测产品的TMB-H是不合适的; (4)TMB值在不同癌种中存在显著差异, 应依据免疫检查点抑制剂(immune check-point inhibitors, ICI)临床疗效确定阈值, 才能最大可能筛选出ICI治疗的潜在获益人群[23, 24]。
由于TMB检测过程中受到诸多因素影响, 包括生物学特性(样本类型, 肿瘤类型等), 分析前因素(样本质量、数量等), 测序因素(DNA捕获区域, panel大小, 富集方法, 测序深度, 测序平台), 生信分析(突变类型, 胚系变异过滤等)及阈值设定等。因此TMB检测标准化面临诸多挑战:产品设计方面, TMB cut-off值的设立及验证?产品预期用途是单药还是联合用药?预测疗效还是预后?标准化方面也有很多困境:不用肿瘤的阈值的标准化; 不同算法的标准化; 性能指标验证方法的可操作性等。因此, 国际上已经建立了TMB基因检测产品的标准化评估体系, 如美国的FoCR(Friends of Cancer Research)项目和欧洲的QuiP(Quality in Pathology)项目。FoCR使用11个实验室检测TMB平台, 不同的TMB评估参数, 为临床研究输出TMB评估推荐规范。FoCR已经发布了第一期研究成果, 通过使用来自32种癌症类型的癌症基因组图谱(The Cancer Genome Atlas, TCGA)的公开数据, 遵循各个检测TMB平台的独特的生物信息学规则算法, 计算出各平台NGS大panel的TMB。发现基于目标区域捕获的NGS大panel与WES具有一定的相关性, 同时对于NGS大panel TMB检测的分析验证也给出了建议[25]。同样, QuiP项目是基于6个NGS大panel, 15个实验中心, 对来自3种肿瘤类型(肺腺癌、头颈部鳞状癌和结肠腺癌)总共20个福尔马林固定石蜡包埋(formalin-fixed paraffin-embedded, FFPE)组织样本进行检测分析。结果显示:16个检测样品在各个实验室间和各个NGS大panel间的TMB变异较低。对比不同实验中心检测TMB的结果, 其中87.7%的检测结果Spearman相关系数大于0.6。NGS大panel检出的TMB和WES的TMB一致率达74.9%。NGS大panel检测TMB主要影响因素包括背景噪音、DNA投入量、测序深度、基因组覆盖区域和检测下限[26]。QuiP提供了TMB检测的真实世界数据, 研究结果提示参与项目的各个NGS大panel及实验平台可用于检测评估TMB, 但需要仔细控制评估TMB的重要参数。FoCR项目与QuiP项目正在努力探索建立TMB检测的规范框架和蓝图。
除了欧美发起的TMB一致性评价项目外, 由中国国家癌症中心中国医学科学院肿瘤医院、中国食品药品检定研究院以及中国抗癌协会肿瘤病理专业委员会联合发起的中国TMB标准化项目(China TMB Harmonization Project)也于2020年10月18日启动, 项目旨在推动中国TMB检测一致性评估与标准化。该项目拟通过对TMB在肿瘤免疫治疗临床应用中所涉及的相关问题进行全面考察和分析, 完成对不同基因检测产品的分析和评估工作; 建立TMB计算方法在不同类型检测方法与WES之间的转换关系; 应用临床试验样本, 结合随访数据, 划定不同癌种免疫治疗的TMB最佳获益基线, 建立TMB临床检测标准及评估体系。项目在方案流程上一定程度地参考了FoCR与QuiP积累的经验, 同时根据中国临床检测的实际情况进行了大量的优化调整。项目总共分为2个阶段, 第一阶段是中国TMB检测现状调研与能力评估, 第二阶段是各癌种TMB临床获益评估与基线建立。
同时还应注意, 对TMB的准确评估(无论基于组织或血浆cfDNA样本)建立在样本满足一定肿瘤占比的基础上, 肿瘤占比过低将导致TMB的严重低估。因此, 临床医生在拿到1份NGS报告、看到TMB数值时, 应综合考虑以下因素:选择的panel(有无准确测算TMB的能力)、检出的肿瘤突变谱及其突变丰度(肿瘤占比是否足以评估TMB、突变谱特征与TMB高/低的组合是否合理)、TMB绝对值以及该数值在已检测的肿瘤样本中的相对排序等, 综合评估TMB水平及其可信度。目前, 国内还没有基于肿瘤组织或血液的NGS大panel产品获批, 不过相信随着NGS大panel检测的标准化发展和建立, 将会有更多的基因检测公司参与到NGS大panel检测产品的注册申报中, 有利于促进中国肿瘤精准诊疗行业的进一步发展。
2.5.2 微卫星不稳定性
微卫星不稳定性(microsatellite instability, MSI)/错配修复(mismatch repair, MMR)检测对于多种实体瘤患者具有重要临床意义。过去以PCR毛细管电泳法作为MSI检测的金标准, 基于美国国家癌症研究所(National Cancer Institute, NCI)推荐的5~7个经典微卫星位点, 对比肿瘤细胞与正常细胞的检测结果, 以确定肿瘤细胞的MSI状态[27]。近年来, NGS panel开始用于MSI检测, 使用计算工具同时研究基因组上的大量微卫星序列成为可能。且多数NGS-MSI算法采用正常人长度分布模型, 无需正常组织作对照。1个NGS panel需要整合一定数量的有效微卫星位点、构建相应的MSI算法并经过临床样本验证, 才能准确报告MSI状态[28]。对MSI的准确评估同样也建立在一定肿瘤占比的基础上。对于MSI检测结果, 应综合样本肿瘤占比(可参考肿瘤突变丰度)、TMB水平[微卫星高度不稳定性(MSI-high, MSI-H)的肿瘤往往TMB-H]、肿瘤突变谱特征(MSI-H可由MMR基因失活性突变导致, 且基因突变谱往往以Indel为主)等信息综合分析, 判断其可信度[29]。
2.5.3 同源重组修复缺陷
同源重组修复缺陷(HRD)通常指细胞水平上的同源重组修复(homologous recombination repair, HRR)功能障碍状态, HRD可由HRR相关基因胚系突变或体细胞突变以及表观遗传失活等诸多因素导致。当HRD存在时, DNA双链断裂会过度依赖非同源末端连接、微同源末端连接和单链退火途径等低保真、高易错的替代性DNA损伤修复途径, 从而极可能造成核酸序列的插入/缺失, 拷贝数异常, 并引起染色体交联, 造成基因组和染色体不稳定。HRD会产生特定的、可量化的、稳定的基因组改变, 可通过建立基于基因组特征分析的评估体系来预测肿瘤HRD状态及其程度, 已成为晚期卵巢癌患者临床应用PARP抑制剂的新型生物标志物, 也可能对乳腺癌、前列腺癌等肿瘤的PARP抑制剂和铂类药物的临床用药具有指导价值[30]。HRD检测采用NGS方法, 通常包括两个部分, BRCA1/2突变状态及基因组不稳定性状态的评分(genomic instability score, GIS), 或称HRD评分(HRD score)。对于后者, 一般通过对细胞内SNP进行检测和计算得出。其原理是基于细胞内因HRD而引起的DNA损伤, 将以一些特定且可识别的方式在基因组上留下痕迹, 如LOH、端粒等位基因失平衡(telomeric allelic imbalance, TAI)和大片段迁移(large-scale state transitions, LST)。截至目前, 全球范围内仅2款HRD检测产品在大型Ⅲ 期临床研究中得到验证:Myriad myChoice® CDx和FoundationFocusTM CDx BRCA LOH。2019年10月, Myriad myChoice® CDx首次被FDA批准作为鉴别HRD阳性的晚期上皮性卵巢癌的伴随诊断; 2020年5月, Myriad myChoice® CDx被FDA批准作为鉴别HRD阳性的晚期卵巢癌患者奥拉帕利一线维持治疗的伴随诊断, 用于筛选奥拉帕利联合贝伐单抗治疗的潜在临床获益人群。而FoundationFocusTM CDx BRCA LOH尚未获批伴随诊断。2020年8月, Myriad myChoice® CDx被ASCO在《PARP inhibitors in the management of ovarian cancer: ASCO guideline》中推荐, 也是唯一提名的商业伴随诊断产品[31]。FDA获批的Myriad myChoice® CDx通过BRCA1/2的致病性变异状态及GIS来评价HRD状态。目前关于Myriad myChoice® CDx的报告解读已有较明确的标准:当BRCA1/2突变和(或)GIS评分≥ 42分时, 即判定为HRD阳性, 详见表4。
| 表4 Myriad myChoice® CDx HRD结果判读 Tab.4 Interpretation of Myriad myChoice® CDx HRD |
最终基于患者HRD状态, 国内外指南及共识的推荐、PARP抑制剂国内外药物适应证, 指导临床合理用药。
3 可报告范围及质量控制
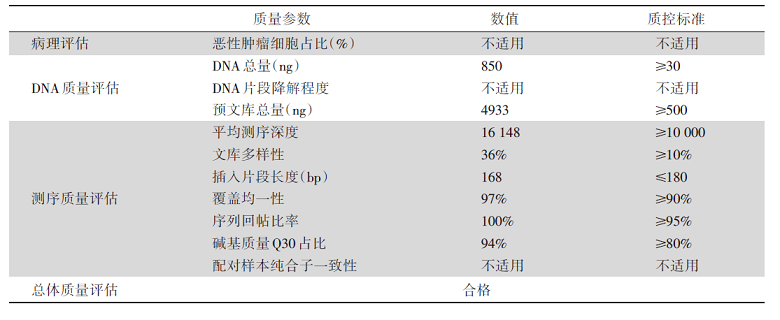
当临床医生拿到1份NGS临检报告时, 应关注其样本主要质控参数、可报告范围(检测内容)、检测方法及其局限性等相关内容, 具体示例详见图3及表5。相关内容可能部分位于报告首页后(如检测内容概述), 而更多详细信息则以附录形式出现在报告主体内容后, 故往往容易被忽略。但1份报告的结果是否真实可信, 或者是否能够回答临床医生最关心的问题, 恰恰严重依赖于这些看似不起眼的“ 附加信息” , 包括检测方法、可报告范围(检测内容)、质控参数、局限性说明等。
 | 图3 NGS报告中对检测方法的说明示例Fig.3 Description of test methods of NGS |
| 表5 NGS报告中的样本主要质控参数示例 Tab.5 Main quality control parameters of sample |
还有一点需要注意, 不同变异形式的检出对样本肿瘤DNA含量的要求是不同的, 换句话说, 在相同的DNA投入量和肿瘤占比的前提下, NGS检测对突变、重排、CNV、TMB及MSI状态的检测敏感性不同。比如1份FFPE样本的肿瘤占比评估约为15%(或maxAF< 10%), 对于基因突变、重排检测及TMB估算来说为合格样本, 但对CNV、MSI的检测敏感性则可能受限。此类信息的获得依赖于对报告“ 局限性说明” 的仔细阅读。
NGS报告端能否遵循恰当的结构化循证原则, 形成可读性较强的临床报告; 临床医生能否正确理解NGS报告的逻辑结构、抓取关键信息并综合分析以指导临床决策, 决定了NGS检测能否为肿瘤患者带来切实的临床获益。无论报告模板如何, 无论遵循哪个具体的变异临床解读逻辑, 任何1份NGS报告的解读结果都不应抵触药物说明书或专业临床指南。临床医生在拿到1份NGS报告后, 不应拘泥于一条条检测报告结果本身, 而应抽提出具体信息, 整合到患者的实际临床背景中去讨论。医学强调循证, 也强调个体化, 临床医生应在综合浏览完1份样本检出的所有分子变异后, 结合患者的基本临床信息以及既往或同期其他配对样本检测结果(如果有), 判断患者疾病的全面分子特征谱及其演化过程, 联系这些信息提示的生物学意义和临床意义后给出临床决策建议, 而不是机械地仅仅基于报告检出的证据级别最高的基因变异给出对应用药推荐。对于复杂情况应鼓励采取多学科讨论(如分子肿瘤专家组, molecular tumor board, MTB)的形式辨析不同变异组合的生物学意义和临床意义[32, 33]。
附录:名词解释
· AF / MAF / VAF(allele fraction / mutation allele fraction / variant allele fraction):突变丰度, 或突变/变异等位基因丰度, 是指某个基因位点所有的等位基因中, 突变的等位基因所占的相对比例, 即等于突变型 / (突变型 + 野生型)。
· CEP(chromosome enumeration probe):染色体计数探针, 能够与位于着丝粒区域高度重复的alpha卫星序列结合, 用于检测染色体的数目异常。
· cfDNA(circulating free DNA):循环游离DNA, 是细胞凋亡或者坏死后降解和释放到外周血的游离DNA。
· CHIP(clonal hematopoiesis of indeterminate potential):意义未明克隆性造血, 是一种普遍的与老化相关的现象, 造血干细胞或其他造血祖细胞形成携带一定基因特征的血细胞亚群。这些亚克隆所携带的变异一般丰度较低, 可以通过白细胞等深度测序对照及生信算法(部分)进行过滤。
· CNV(copy number variation):拷贝数变异, 是基因组上部分区域的扩增或缺失。CNV是一种结构变异, 在肿瘤基因组中常见, 包括插入、缺失和重复。NGS可通过对该区域的覆盖度(测序深度)来估算CNV。
· ctDNA(circulating tumor DNA):循环肿瘤DNA, 是外周血中携带一定肿瘤特征(如SNV、插入/缺失突变、重排、CNV等), 来自肿瘤基因组的DNA片段。主要来源有凋亡或坏死的肿瘤细胞、循环肿瘤细胞(circulating tumor cell, CTC)以及肿瘤细胞分泌的外泌体。
·HRD(homologous recombination deficiency):同源重组修复缺陷, 通常指细胞水平上的同源重组修复(homologous recombination repair, HRR)功能障碍状态, HRD可由HRR相关基因突变以及表观遗传失活等诸多因素导致。
· LBx(liquid biopsy):液体活检, 是指对非固体生物样本(主要是血液)的采样和分析。液体活检和组织活检一样, 主要用于癌症等疾病的诊断和监测, 与组织活检相比具有无创, 能动态检测等优点。
· LOD(limit of detection):最低检测限, 是指能以适当的置信度(一般是95%)被检出的突变的最低浓度。
· LOH(loss of heterozygosity):杂合性缺失, 是一种使某特定基因丢失的基因组变化。如果一个特定位点的等位基因一侧正常、一侧有突变, 某种原因导致的正常等位基因缺失或突变会产生没有正常功能的基因座。
· MaxAF / MSAF(maximum allele fraction /
maximum somatic allele frequency):最大变异丰度, 或最大体细胞基因变异频率, 是指在组织、血浆等类型样本中, 所检出的最高的体细胞变异丰度。
· MMR(mismatch repair):错配修复。DNA错配修复是指在含有错配碱基的DNA分子中, 使核苷酸序列恢复正常的一种修复方式。
· MRD(minimal/molecular/measurable residual disease):微小残留病灶, 分子残留病灶或可测量残留病灶。其概念源于白血病在诱导化疗达到完全缓解(或骨髓移植治疗)后, 在患者体内依然残留着痕量白血病细胞的状态。在实体瘤的应用中, 泛指在根治性治疗(如手术切除)后, 传统影像学或实验室方法不能发现病灶, 但通过液态活检技术可检出癌细胞来源分子的情况。
· MSI(microsatellite instability):微卫星不稳定性。微卫星是指细胞基因组中以少数几个核苷酸(多为1~6个)为单位串联重复的DNA序列。DNA错配修复(MMR)功能出现异常时, 微卫星出现的复制错误得不到纠正并不断累积, 使得微卫星序列长度或碱基组成发生改变, 称为微卫星不稳定性。
· SNV(single-nucleotide variant):单核苷酸变异, 是基因组上部分区域发生的点突变。SNV是肿瘤基因组变异最常见的类型。
· TBx(tissue/tumor biopsy):组织活检或肿瘤组织活检, 是采集组织并对组织进行检查和分析。
· TMB(tumor mutational burden):肿瘤突变负荷, 是指肿瘤基因组编码区每百万碱基中的体细胞非同义突变个数。理论上TMB越高, 可能被T淋巴细胞识别的新抗原越多, 免疫检查点抑制剂疗效可能越好。
解读肿瘤学专家组成员
名誉主编:吴一龙(广东省人民医院)
执笔组长:张绪超(广东省人民医院)
执笔委员会成员(按姓名拼音排序):
蔡林波(广东三九脑科医院), 常建华(中国医学科学院肿瘤医院深圳分院), 陈锋夏(海南省人民医院), 陈炬(中山大学孙逸仙纪念医院), 成建德(山西白求恩医院), 关印(首都医科大学附属北京朝阳医院), 郭卉(西安交通大学第一附属医院), 郭禹标(中山大学附属第一医院), 黄媚娟(四川大学华西医院), 蒋华(常州市第二人民医院), 金时(中国医学科学院肿瘤医院深圳医院), 李敏(中南大学湘雅医院), 李延(四川省人民医院), 李子明(上海交通大学附属胸科医院), 梁红玲(广州医科大学附属肿瘤医院), 柳江(新疆维吾尔自治区人民医院), 陆元志(暨南大学附属第一医院), 罗永忠(湖南省肿瘤医院), 马虎(遵义医科大学第二附属医院), 马建群(哈尔滨医科大学附属肿瘤医院), 潘小杰(福建省立医院), 秦建文(天津市胸科医院), 邱剑光(中山大学附属第六医院), 斯璐(北京大学肿瘤医院), 宋子正(河北大学附属医院), 孙玉萍(山东省肿瘤医院), 涂海燕(广东省人民医院), 王慧娟(河南省肿瘤医院), 吴迪(深圳市人民医院), 吴现瑞(中山大学附属第六医院), 谢展鸿(广州医科大学附属第一医院), 徐蓓(复旦大学附属中山医院), 许隽颖(无锡市人民医院), 杨学宁(广东省人民医院), 姚文秀(四川省肿瘤医院), 叶峰(厦门大学附属第一医院), 叶贤伟(贵州省人民医院), 岳东升(天津医科大学肿瘤医院), 岳君秋(湖北省肿瘤医院), 臧远胜(海军军医大学附属长征医院), 张红梅(空军军医大学西京医院), 张俊(上海交通大学医学院附属瑞金医院), 张璐(广州燃石医学检验所有限公司), 张晓春(青岛大学附属医院), 张新伟(天津医科大学肿瘤医院), 张永昌(湖南省肿瘤医院), 张瑜(中南大学湘雅医院), 张周(广州燃石医学检验所有限公司), 赵宏(中国医学科学院肿瘤医院), 赵军(苏州大学附属第一医院), 周建娅(浙江大学医学院附属第一医院), 庄莉(云南省肿瘤医院)
顾问委员会成员(按姓名拼音排序):
卜庆(广西医科大学第一附属医院), 陈功(中山大学附属肿瘤医院), 陈莉莉(台州市第一人民医院), 陈理明(汕头大学医学院第一附属医院), 陈鸰(中南大学湘雅医院), 陈新(南方医科大学珠江医院), 陈宇(广东省人民医院), 陈元(华中科技大学同济医学院附属同济医院), 陈志康(中南大学湘雅医院), 褚倩(华中科技大学同济医学院附属同济医院), 崔健(北京市垂杨柳医院), 崔久嵬(吉林大学第一医院), 崔同建(福建省立医院), 丁浩(镇江市第一人民医院), 丁礼仁(浙江大学医学院附属第二医院), 董宇超(海军军医大学第一附属医院), 杜瀛瀛(安徽医科大学第一附属医院), 段萍(成都市第一人民医院), 范云(浙江省肿瘤医院), 范晓溪(中国医科大学附属第一医院), 耿庆(武汉大学人民医院), 古卫权(佛山市第一人民医院), 郭人花(江苏省人民医院), 郝静(山东大学齐鲁医院), 何勇(陆军特色医学中心), 侯小明(兰州大学第一医院), 胡文蔚(常州市第一人民医院), 黄镜(中国医学科学院肿瘤医院), 季有信(青岛市中心医院), 姜国忠(郑州大学第一附属医院), 蒋军红(苏州大学附属独墅湖医院), 李明哲(中山大学附属第七医院), 李显桃(益阳市中心医院), 李智(广东省人民医院), 黎扬斯(广东省人民医院), 梁乃新(北京协和医院), 林根(福建省肿瘤医院), 林丽珠(广州中医药大学第一附属医院), 刘思阳(暨南大学附属第一医院), 刘思旸(广东省人民医院), 刘先领(中南大学湘雅二医院), 刘洋(四川省肿瘤医院), 刘莺(河南省肿瘤医院), 吕冬青(台州恩泽医疗中心台州医院), 马杰(河南省肿瘤医院), 孟宏学(哈尔滨医科大学附属肿瘤医院), 欧阳能太(中山大学孙逸仙纪念医院), 潘跃银(安徽省肿瘤医院), 乔贵宾(广东省人民医院), 饶创宙(中国科学院大学宁波华美医院), 单建贞(浙江大学医学院附属第一医院), 宋霞(山西省肿瘤医院), 宋勇(东部战区总医院), 宋正波(中国科学院大学附属肿瘤医院), 孙浩(重庆大学附属肿瘤医院), 唐源(四川大学华西医院), 王刚(唐山开滦总医院), 王根和(黄山市人民医院), 王洪琰(河北医科大学第四医院), 王继勇(广州中医药大学第一附属医院), 王佳蕾(复旦大学附属肿瘤医院), 王珂(天津医科大学肿瘤医院), 王琳琳(山东省肿瘤医院), 王晰程(北京大学肿瘤医院), 王晓东(北京大学首钢医院), 王燕(中国医学科学院肿瘤医院), 王志杰(中国医学科学院肿瘤医院), 魏慎海(清华大学第一附属医院), 温灏(复旦大学附属肿瘤医院), 温昭科(广西壮族自治区人民医院), 邬麟(湖南省肿瘤医院), 项轶(上海交通大学医学院附属瑞金医院), 肖德胜(中南大学湘雅医院), 肖海平(广东药科大学附属第一医院), 辛华(吉林大学中日联谊医院), 徐嵩(天津医科大学总医院), 徐艳珺(浙江省肿瘤医院), 徐智(陆军军医大学第二附属医院), 杨帆(北京大学人民医院), 杨华(河北大学附属医院), 杨衿记(广东省人民医院), 杨林(深圳市人民医院), 杨农(湖南省肿瘤医院), 杨宇(哈尔滨医科大学附属第二医院), 姚煜(西安交通大学第一附属医院), 殷伟强(广州医科大学附属第一医院), 虞巍(北京大学第一医院), 袁瑛(浙江大学医学院附属第二医院), 曾珊(中南大学湘雅医院), 张沂平(中国科学院大学附属肿瘤医院), 张智弘(江苏省人民医院), 赵光强(云南省肿瘤医院), 赵军(北京大学肿瘤医院), 赵明芳(中国医科大学附属第一医院), 赵晓亮(天津医科大学肿瘤医院), 钟华(上海交通大学附属胸科医院), 钟文昭(广东省人民医院), 周蓉蓉(中南大学湘雅医院), 周承志(广州医科大学附属第一医院), 周清(广东省人民医院), 周晓燕(复旦大学附属肿瘤医院), 周永春(云南省肿瘤医院), 朱梁军(江苏省肿瘤医院), 朱耀(复旦大学附属肿瘤医院), 宗红(郑州大学第一附属医院)
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|