
{kind=link}
{kind=link}
{kind=link}
原发性皮肤T细胞淋巴瘤患者的临床特征及预后分析
[聂琳琳1 , 董舒蕾1 , 李开然1 , 李君1 , 江玲玲1 , 沈汉影1 , 朱锋2  ]
]
]
|
|


聂琳琳(1991-),女,河南驻马店人,在读硕士研究生,主要研究方向为淋巴瘤。
目的 探讨原发性皮肤T细胞淋巴瘤(cutaneous T-cell lymphoma,CTCL)的临床特征、治疗方式及预后。方法 回顾性分析徐州医科大学附属医院2009年11月至2019年5月经皮肤活检确诊为原发性CTCL患者15例,以描述这种罕见肿瘤。结果 确诊患者中,男性8例,女性7例,中位年龄50岁,绝大多数患者(13/15)因皮肤病变就诊。各亚型的CTCL皮损形态各异。蕈样肉芽肿(mycosis fungoides,MF)主要表现为斑块(83.3%);皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma,SPTCL)主要表现为肢体多发性皮下结节(75%);外周T细胞淋巴瘤非特指型(peripheral T-cell lymphoma not otherwise specified,PTCL-NOS)皮损呈多态性;主要皮损部位为肢体(7例)和全身(3例)。各亚型的CTCL临床病理特征不同。瘤细胞表面以T细胞抗原表达为主,Ki-67阳性指数5%~80%,1例EB病毒编码区(Epstein-Barr encoding region,EBER)为(+)。中位随访时间为54.3个月,2年总生存率(overall survival,OS)为66.7%,2年无进展生存率(progression-free survival,PFS)为43.9%。7例死亡患者中位生存时间为18个月(14.0~57.2个月)。惰性或低危组患者的4年OS( P=0.045 4)和PFS( P=0.002 3)显著高于侵袭性或高危组,MF、SPTCL、PTCL-NOS之间的PFS有显著差异( P=0.033 4)。结论 本研究中,除MF或SPTCL外,其余CTCL亚型侵袭性强,预后差。各亚型皮损表现及组织病理形态多样化,诊断时应结合临床表现、组织病理检查、免疫表型等,以免漏诊、误诊。
Objective To investigate the clinical features, treatment, and prognosis of primary cutaneous T-cell lymphoma (CTCL).Methods Totally 15 patients with primary CTCL diagnosed by skin biopsy in the affiliated Hospital of Xuzhou Medical University from November 2009 to May 2019 were retrospectively analyzed to describe this rare tumor.Results The main results were as follows: 8 were males and 7 were females, and the median age was 50 years old. The vast majority of patients (13/15) see a doctor for skin lesions. The skin lesions of different subtypes of CTCL vary in shape. The main manifestation of mycosis fungoides (MF) was plaque (83.3%); Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) was mainly characterized by multiple subcutaneous nodules of the extremities (75%); peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) lesions were polymorphic. The main lesions were limb ( n=7) and whole body ( n=3). The clinicopathological features of different subtypes of CTCL were different. T cell antigen was mainly expressed on the surface of tumor cells. The positive index of Ki-67 was 5%~80%. Epstein-Barr encoding region (EBER) was positive in only 1 case. The median follow-up time was 54.3 months, 2-year overall survival (OS) was 66.7%, and 2-year progression-free survival (PFS) was 43.9%. The median survival time of 7 dead patients was 18 months (range 14.0~57.2). The 4-year OS ( P=0.045 4) and PFS ( P=0.002 3) of patients in the indolent or low-risk group were significantly better than those in the aggressive or high-risk group, and there were significant differences in PFS among MF, SPTCL, and PTCL-NOS ( P=0.033 4).Conclusions In this study, except for MF or SPTCL, the other CTCL subtype had strong invasiveness and poor prognosis. The manifestations and histopathological morphology of various subtypes of skin lesions were diversified. Clinical manifestations, histopathological examination, and immunophenotype should be combined in the diagnosis to avoid missed diagnosis and misdiagnosis.
原发性皮肤T细胞淋巴瘤(cutaneous T cell lymphoma, CTCL)是以皮肤相关T淋巴细胞恶性增殖为基础的一组异质性疾病, 是第二大结外非霍奇金淋巴瘤(non-Hodgkin lymphoma, NHL), 同时也是全球范围内预后较差的肿瘤之一。不同亚型的CTCL具有不同的组织学和免疫组织学特征, 其临床表现、病程预后也各异。随着对CTCL基本认识的提高, 世界卫生组织-欧洲癌症研究与治疗组织(World Health Organization-European Organization for Research on Treatment of Cancer, WHO-EORTC)于2005年对其提出了分类, 并于2018年进行更新[1, 2]。由于该病是一种极其罕见的血液系统恶性肿瘤, 检索文献多为少数病例的报道, 为提高对该组疾病的认识, 增加诊断及鉴别诊断思路, 本研究回顾性分析了我们中心诊治的15例患者资料, 总结其临床特点、治疗及预后。
收集2009年11月至2019年5月在徐州医科大学附属医院诊治的15例CTCL患者, 参照2018年WHO-EORTC皮肤淋巴瘤分类标准进行分类[2], 并依据2005年WHO-EORTC皮肤淋巴瘤分类建立风险组[1]。入选标准:(1)诊断时无皮外受累, 且病理学证实为原发性CTCL患者; (2)正式治疗前完善相关检查后符合入组条件; (3)截至随访时间至患者疾病稳定或复发或死亡。排除标准:(1)临床资料不完整, 且确诊后于我院治疗< 2疗程; (2)外周T细胞淋巴瘤其他亚型; (3)因全身疾病或其他原发部位肿瘤继发皮肤受累的患者。收集有关年龄、性别、疾病类型、病理学资料、B症状(连续3天发烧, 6个月体重下降10%以上, 盗汗)、淋巴结病变、皮肤受累范围、乳酸脱氢酶(lactate dehydrogenase, LDH)水平、血清EB病毒水平、β 2微球蛋白水平、治疗方案等信息。
所有患者均接受详细的体格检查、实验室检查、影像学检查[胸部计算机断层扫描(computed tomography, CT)、腹部B超或CT、全身浅表淋巴结B超检查]; 部分患者接受全身正电子发射断层扫描(positron emission tomography, PET)/CT检查。蕈样肉芽肿(mycosis fungoides, MF)/塞扎里综合征(Sé zary syndrome, SS)的分期参照国际皮肤淋巴瘤学会(International Society for Cutaneous Lymphomas, ISCL)和EORTC更新标准[3], 非MF/SS的分期标准参照文献[4]。
根据WHO疗效评价标准进行判定, 疗效分为完全缓解(complete response, CR)、部分缓解(partial response, PR)、疾病稳定(stable disease, SD)和疾病进展(progressive disease, PD)。
采用电话随访的形式, 随访起始为病理确诊之日, 截止日期2022年3月31日。无进展生存(progression-free survival, PFS)时间定义为从接受治疗开始至发生进展或复发, 或发生因任何病因死亡或末次随访时间。总生存(overall survival, OS)时间定义为从确诊时间至因任何原因死亡的时间或末次随访截止时间。两者均以月为单位。
采用GraphPad Prism 8进行统计学分析。采用Kaplan-Meier法绘制生存曲线, 用Log-rank检验进行生存相关预后危险因素分析, P< 0.05有统计学意义。
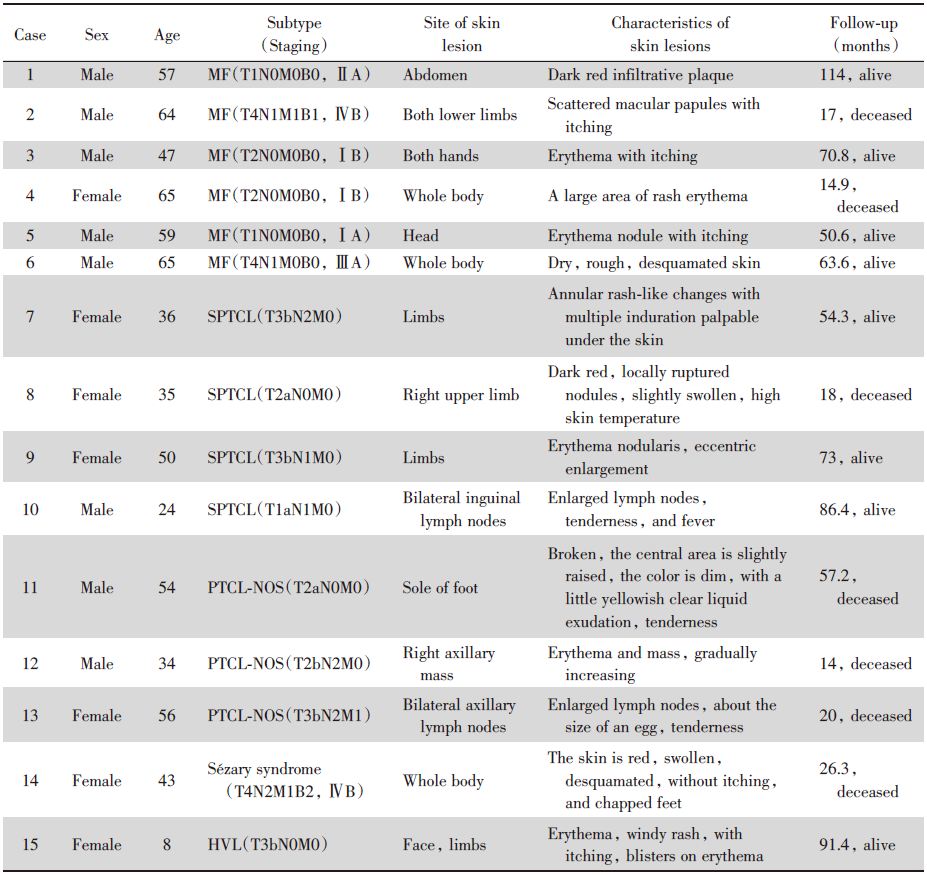
15例患者中, 男性8例, 女性7例, 男女比例为1.1∶ 1.0, 中位年龄50岁(8~65岁), 50~59岁年龄组发病率最高。15例患者的临床特征见表1, 频数分布表见表2。13例患者因皮肤病变入院就诊, 其余患者因淋巴结肿大就诊。5例呈大小不一结节性红斑, 部分表面溃破; 3例肢体多处皮肤结节, 伴瘙痒; 2例表现淋巴结肿大(1例双侧腹股沟淋巴结肿大伴颜面部及下肢水肿, 1例双侧腋窝淋巴结肿大后进展至多部位淋巴结肿大); 2例表现大片皮疹, 高于皮肤, 融合成片, 表面脱屑; 1例风团样皮疹, 局部伴有水疱, 消退后遗留痘疮样瘢痕; 1例皮肤溃疡, 创面红肿、破溃、渗出; 1例右腋下24 cm× 23 cm巨大肿块。皮损分布在肢体7例, 全身3例, 淋巴结2例, 躯干、头部、腋下各1例。26.7%的病例为播散性皮肤受累, 73.3%的病例局限于解剖区域。
| 表1 15例原发性皮肤T细胞淋巴瘤患者的临床资料 Tab.1 Clinical data of 15 patients with primary cutaneous T-cell lymphoma |
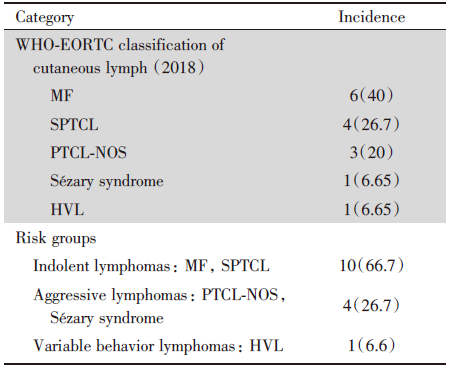
| 表2 原发皮肤T细胞淋巴瘤的发生率和危险组 Tab.2 Primary cutaneous T-cell lymphomas frequency and risk group n(%) |
确诊前皮损持续时间从1个月到47个月不等, 但通常很短(中位9个月)。疼痛和瘙痒的发生率分别为46.7%和26.7%。初诊时, 46.7%(7/15)的患者出现B症状, 53.3%(8/15)的患者LDH水平升高, 46.7%(7/15)的患者β 2微球蛋白升高。1例HVL患者血清EB病毒异常升高。
6例MF患者中, 3例接受干扰素联合黑光治疗, 1例行干扰素联合CHOP(环磷酰胺 + 长春地辛 + 表柔比星 + 泼尼松)化疗, 1例行干扰素辅以VDCP(环磷酰胺 + 长春地辛 + 柔红霉素 + 泼尼松)化疗联合鞘内注射化疗药物(甲氨蝶呤、地塞米松、阿糖胞苷), 1例行CHOP联合MINE(异环磷酰胺、依托泊苷、盐酸米托蒽醌)方案挽救化疗; 4例SPTCL、1例Sé zary综合征和1例HVL患者均接受基于CHOP方案为主的综合化疗, 其中1例SPTCL患者于6周期化疗后行自体造血干细胞移植(autologous hematopoietic stem cell transplantation, ASCT)达CR; 3例PTCL-NOS患者中, 1例行单纯CHOP方案化疗, 1例行CHOP方案联合放疗及西达苯胺靶向治疗, 1例行GDP(环磷酰胺 + 长春地辛 + 地塞米松)方案化疗。全组患者中位化疗6(2~15)个周期。
组织学上, 多数病变表现瘤细胞侵犯表皮, 可见皮肤附属器累及, 真皮浸润, 伴有血管及管周破坏, 部分可累及皮下脂肪组织。细胞形态学上, 多数为小至中等淋巴细胞混合。免疫组化, 所有病例瘤细胞表达CD3, 4例SPTCL患者表达CD8、T细胞胞质内抗原1(T cell intracellular antigen 1, TIA-1)、颗粒酶B(granzyme B, GrB), Ki-67阳性指数5%~80%, 1例HVL患者原位杂交检出EB病毒阳性, 1例CD56阴性病例发现T细胞受体克隆性重排。
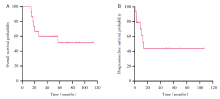
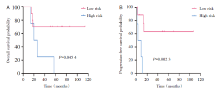
15例患者中, 中位随访时间54.3个月(14~114个月), 中位OS为54.3个月, 中位PFS为10.8个月, 2年OS为66.7%, 2年PFS为43.9%(见图1)。惰性或低危组(MF、SPTCL)患者的OS(4年OS:70% vs. 25%, P=0.045 4)和PFS(4年PFS:63.5% vs. 0%, P=0.002 3)显著优于侵袭性或高危组(PTCL-NOS、Sé zary综合征)(见图2)。如图3所示, MF、SPTCL和PTCL-NOS的4年OS分别为66.7%、75%和33.3%, 4年PFS分别为60%、66.7%和0%。至随访结束, 8例患者存活, 7例患者死亡。其中6例患者(40%)处于完全、长期(≥ 18个月)缓解, 分别是3例早期MF, 2例SPTCL, 1例HVL。
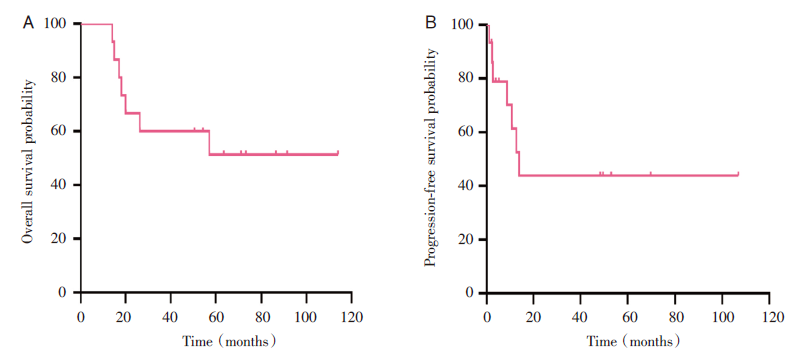 | 图1 15例原发性CTCL的无进展生存和总生存情况 注:A, 总生存; B, 无进展生存。Fig.1 Progression free probability and overall survival in 15 cases of primary CTCL Note: A, Overall survival; B, Progression free probability. |
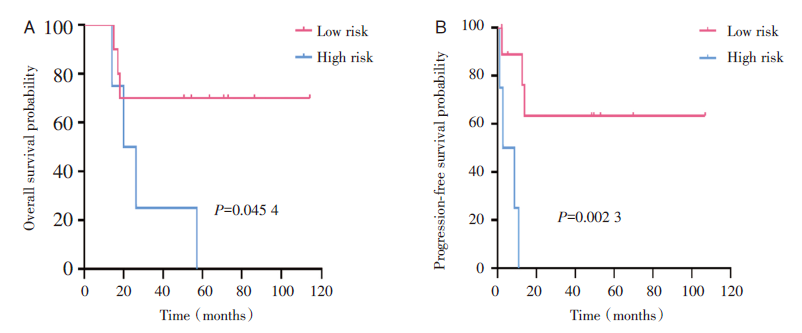 | 图2 根据WHO-EORTC分类按危险组划分的总生存率和无进展生存率(2005) 注:A, 总生存; B, 无进展生存。Fig.2 Overall survival and progression-free survival by risk groups according to the WHO-EORTC classification (2005) Note: A, Overall survival; B, Progression-free probability. |
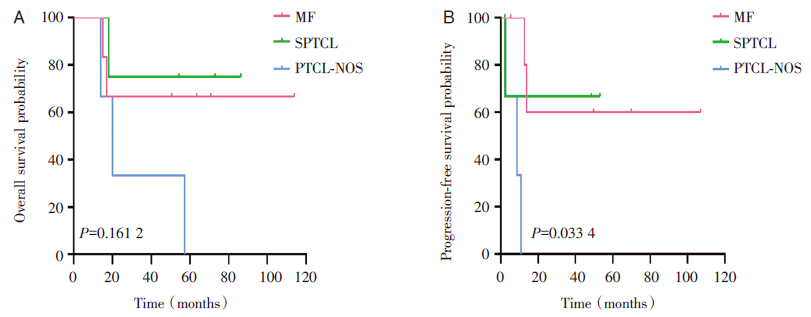 | 图3 MF、SPTCL和PTCL-NOS的总生存率和无进展生存率 注:A, 总生存; B, 无进展生存。Fig.3 Overall survival and progression-free survival for MF, SPTCL and PTCL-NOS Note: A, Overall survival; B, Progression-free probability. |
原发性皮肤淋巴瘤(primary cutaneous lymphoma, PCL)是一组原发于皮肤的异质性NHL, 占结外NHL的18%, 皮肤是仅次于胃肠道的第二常见部位[5, 6]。与其他NHL相比, PCL患者数量较少, 亚型数量较多。根据肿瘤细胞的来源, 可分为CTCL(占PCL的75%~80%)和皮肤B细胞淋巴瘤(占PCL的20%~25%)[7]。鉴于CTCL所占比重高, 本文围绕CTCL进行展开, 以提高临床医师对其诊疗的进一步认识。
CTCL中最常见的类型是MF, 约占CTCL患者总数的50%~70%, 占所有PCL的近50%[1]。Ⅰ A~Ⅱ A期是MF的早期阶段, 所有国内和国际的皮肤淋巴瘤指南推荐这类患者首先进行光疗法。其次, resiquimod是Toll样受体7和8(Toll-like receptor 7, TLR7/TLR8)的激动剂, 在Ⅰ 期研究中显示出对早期MF病例的高应答率[8]。Ⅱ B~Ⅳ B期为MF的晚期, 指南统一建议开始系统治疗, 并结合或不结合皮肤局部治疗, 然而免疫疗法似乎明显优于化疗。维布妥昔单抗(brentuximab vedotin, BV)为靶向CD30的抗体偶联药物, 在多中心研究中显示出在早晚期CTCL中的显著疗效, 于2017年被批准用于CD30阳性CTCL[9]。同样, 专门针对CTCL恶性肿瘤细胞上特异表达KIR3DL2受体的lacutamab也显示了类似的抗晚期肿瘤效应[10]。本研究6例MF患者, 3例行非化疗, 2例存活, 1例死于其他疾病; 3例行系统性化疗, 2例存活, 1例Ⅳ B期患者因肿瘤细胞侵犯骨髓转归至淋巴瘤白血病, 后累及中枢神经系统而死亡。可见, 从治疗的角度来看, MF的惰性和分期是重要的。
SPTCL是一种较罕见的CTCL, 以表达α /β T细胞受体的CD8+ T细胞浸润皮下脂肪组织为特征。早在2005年WHO-EORTC分类中, 将其与更具侵袭性的原发性皮肤γ δ T细胞淋巴瘤(cutaneous gamma/delta T-cell lymphoma, CGDTCL)区分开来。Willemze等[11]发现SPTCL主要累及皮下组织, CD4-、CD8+、CD56-、β F1+, 5年OS为82%。17%的患者继发嗜血细胞综合征(hemophagocytic syndrome, HPS), 且无HPS患者的存活率显著高于HPS患者(5年OS:91% vs. 46%, P< 0.001)。与SPTCL不同, GGDTCL通常累及真皮和表皮, CD4-、CD8-、CD56+/-、β F1-, 5年OS仅为11%。本中心4例患者均为成人, 女性多见, 中位年龄35.5岁, 3例表现为累及肢体和躯干的多发性皮下结节, 1例累及腹股沟, 这一结果与之前在SPTCL其他系列报告的一致[11, 12, 13]。在早期一般无明显淋巴结受累, 但在1/3初诊病例和绝大多数病例的病程中会继发HPS, 导致患者死亡, 这与本中心1例35岁年轻患者继发HPS结局一致, 多项研究也支持继发HPS会导致高致死率[11, 14]。目前, SPTCL常规采用以阿霉素为基础的多药化疗, 但也有研究推荐免疫抑制剂应作为一线治疗药物[15]。有趣的是, 本中心1例患者在以阿霉素为主的综合化疗后病情进展, 挽救治疗中靶向应用西达苯胺达到CR, 可见新药为SPTCL带去了希望。
目前认为Sé zary综合征代表CTCL的白血病阶段, 是侵袭性较强的CTCL。患者外周血出现异常单一核细胞, 即Sé zary细胞。但检测外周血中Sé zary细胞的计数不是CTCL特有的, 因为在健康人和良性疾病的外周血中可能发现Sé zary细胞[16]。CD7和/或CD26缺失对其诊断具有敏感性(> 80%)和高度特异性(100%)[17]。Sé zary综合征患者淋巴细胞亚群变化显著, 尤其CD4/CD8≥ 10。2017年EORTC更新的治疗指南分为一线和二线方案[18]。前者包括体外光化学疗法、苯丁酸氮芥+强的松、体外光化学疗法或8-甲氧补骨脂素加紫外线-A(ultraviolet-A, UVA)联合全身治疗、类维生素A、干扰素-a、小剂量甲氨蝶呤(methotrexate, MTX)。后者多为CHOP或类CHOP化疗, 还有免疫调节药(阿仑单抗)、异基因干细胞移植。莫格利珠单抗(mogamulizumab)是第一个靶向碳分解代谢产物阻遏4(carbon catabolite repression 4, CCR4)的生物制剂, 尤适用于有血液受累的Sé zary综合征患者[19]。本中心仅有1例Sé zary综合征患者, 其CD4/CD8=97, CD7-, CD26-, 血片观察见异常Sé zary细胞, 该细胞中等大小, 核深染, 核型不规则伴有脑回样等畸形改变, 符合ISCL Sé zary综合征的诊断标准[20]。初诊时白细胞68.4× 109/L, 抗核抗体(antinuclear antibody, ANA)阳性, 骨穿提示幼稚淋巴细胞白血病, FISH(BCR-ABL):阳性细胞数10, 尽管先后予以CHOP、维A酸、MTX等综合治疗, 仍于确诊后26.3个月死亡, 可见其恶性程度高。
PTCL-NOS不能归入原发性CTCL中任一明确类型, 它可能包含多种亚型, 目前临床尚缺乏识别它们的特异性生物标志物。不论所属何种分期, 首先推荐参加合适的临床试验, 或4~6个周期化疗± 放疗。对于治疗后达CR的患者, 可继续参加合适的临床试验、ASCT甚至停药观察。最新研究显示肿瘤细胞表面mTOR突变可能代表了一个新的治疗靶点[21]。一项对82例PTCL-NOS患者的生存分析显示, 接受放疗的31例患者有22例(71%)获得CR, 接受阿霉素为主化疗的36例患者有10例(28%)获得CR, 但达CR患者的中位无病生存期仅为8个月。中位随访19个月, 仅13例患者存活, 总的5年生存率仅为20%[22]。本研究中, 他们占病例的20%, 多采用基于阿霉素为主的化疗, 尽管在复发后积极应用二线方案(西达苯胺、GDP)挽救治疗, 3例患者均死亡, 2年OS为33.3%。可见该病侵袭性强, 预后极差。
区别于其他CTCL, HVL与慢性活动性EBV感染高度相关, 有发展为淋巴瘤的风险。有学者提出[23], 紫外线可以诱发或加重皮肤损害, 因此该类患者应避免阳光下暴晒。Verneuil等报道称[24], 血清EBV DNA载量在疾病活动期增加。多项研究表明[25, 26], 伴有NK细胞表型(如CD56+)的患者比T细胞表型的患者病程更缓慢。Sangueza等回顾性分析了南美12例接受化疗辅以放疗的HVL患者[27], 8例患者在诊断后平均5.3个月死亡, 提示化疗可能与HVL患者预后不良有关。本中心收集的这一例8岁儿童在出现新发水疱时血清EBV-DNA载量显著升高, 且EBER(+), 免疫组化提示CD56+, 经沙利度胺联合CHOP方案辅以抗病毒治疗后EBV下降明显, 至随访结束仍存活, 表明HVL患者采用该方案可能延长患者生存期。
综上, 原发性CTCL是一组异质性的罕见疾病, 除MF、原发性皮肤CD30+淋巴增生性疾病和SPTCL外, 其他亚型更具侵袭性, 且现有的治疗方案中达永久性完全缓解很少见, 提示原发性CTCL的诊治仍存在挑战。本研究揭示惰性淋巴瘤的预后好于侵袭性淋巴瘤, 并描述了各亚型的临床特征, 为临床医师更好的认识该病种提供了借鉴与参考。但本文系小样本研究, 患者数量有限且研究具有回顾性, 以后将继续积累病例, 以期为临床诊治提供帮助。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|