{kind=link}
蛋白酪氨酸磷酸酶SHP2在非小细胞肺癌中的靶向治疗研究进展
[周文斌1, 2 , 张绪超1, 2  ]
]
]
|
|


周文斌(1997-),女,湖南衡阳人,在读硕士研究生,主要研究方向为肺癌肿瘤微环境。
由PTPN11编码的非受体含Src同源2结构域的蛋白酪氨酸磷酸酶(Src homology 2 domain containing protein tyrosine phosphatase,SHP2),是肿瘤细胞中多条信号通路的汇聚节点。SHP2参与了多种不同恶性肿瘤的发病机制,这其中也包括了非小细胞肺癌(non-small cell lung cancer,NSCLC)。SHP2参与调节肿瘤细胞的增殖、凋亡、侵袭及转移过程等。在多项肿瘤治疗后耐药的模型中,联合使用SHP2抑制剂与其他治疗手段可以使耐药肿瘤消退,重塑肿瘤微环境。本文综述了SHP2在NSCLC中的生物学功能,以及SHP2抑制剂的开发和其在NSCLC中的应用,展现了SHP2靶点在NSCLC中的治疗前景。
The non-receptor Src homology 2 domain-containing protein tyrosine phosphatase (SHP2) was encoded by PTPN11, converging multiple signaling pathways in tumor cells. SHP2 was involved in the pathogenesis of many different malignancies, including non-small cell lung cancer (NSCLC). SHP2 regulated cell proliferation, apoptosis, invasion, and metastasis in lung cancer. In several models of therapeutic resistance, the combination of SHP2 inhibitors with other therapies can regress resistant tumors and remodel the tumor microenvironment. This paper reviews the biological functions of SHP2 in NSCLC, and summarizes the development of SHP2 inhibitors and their applications in NSCLC, showing the great potential of SHP2 as a drug target for NSCLC.
蛋白质磷酸化是一种广泛存在的蛋白质翻译后修饰, 几乎参与细胞所有的生命活动, 是细胞生命活动上重要的调控开关。蛋白质的酪氨酸磷酸化是由蛋白质酪氨酸磷酸激酶(receptor tyrosine kinase, RTKs)及蛋白质酪氨酸磷酸酶(protein tyrosine phosphatase, PTPs)介导调控的。而在人类基因组107个PTP基因中, 至少有一半在免疫系统也有表达[1]。含Src同源2结构域的蛋白酪氨酸磷酸酶(Src homology 2 domain containing protein tyrosine phosphatase, SHP2)是在细胞内普遍表达的一种酪氨酸磷酸酶。在RAS/ERK通路中, SHP2位于RAS的上游, 是癌症通路中的重要信号级联节点, 几乎所有的RTKs激活RAS/ERK通路都需要激活SHP2[2]。除此以外, SHP2也是JAK/STAT、NF-κ B和PI3K/AKT通路的关键调控因子之一[3, 4, 5, 6]。SHP2在肺癌的发生发展中也起到了重要作用[7], 因此, SHP2是一个非常有前景的肺癌靶点, SHP2抑制剂在肺癌中的应用策略需要更多的临床试验来探索。
SHP2在美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)基因数据库中的官方名称为蛋白质酪氨酸磷酸酶非受体型11(protein tyrosine phosphatase non-receptor type 11, PTPN11), 由593个氨基酸组成, 包含两个Src同源2结构域(N-SH2和C-SH2)和一个PTP催化域[8]。在非活性状态下, SHP2的N-SH2结构域与PTP结构域相结合会阻断其配体进入活性位点, 保持在自抑制构象中无法发挥磷酸酶的作用。当SHP2与特定的磷酸酪氨酸pTyr基序(如受体酪氨酸激酶、支架蛋白受体、细胞因子受体)结合时, 蛋白构象发生变化, N-SH2结构域远离PTP域, 使SHP2的自抑制状态被破坏变为开放构象, 从而激活SHP2发挥去磷酸化的作用[8, 9, 10]。SHP2的活化会使Ras持续过度激活以及ERK1/2和AKT途径同时激活, 导致恶性肿瘤的发生; 而SHP2信号通路的失调可能会引起发育障碍。因此, 由于SHP2具有双重作用, 其激活状态也会受到精准的调控[11]。
SHP2在肿瘤中可以检测到不同突变体, 其激活突变与肿瘤的发生有关。SHP2的激活突变体主要可以分为自抑制解除型(E76K), 以及催化活性消除型(C459E)[12]。SHP2的大多数突变体是由N-SH2和PTP结构域的突变形成的。这些突变可以影响SHP2的分子内相互作用, 导致SHP2的激活[13, 14]。SHP2在肺癌中常见的突变体有N-SH2域的V45L、N58S、E76V[15]。近期一项研究对28 686例中国肺癌患者进行回顾性分析, 发现编码SHP2的PTPN11在中国肺癌患者中突变频率仅为0.67%, 其中最常见的突变是G503V。PTPN11也常与RTKs家族成员表皮生长因子受体(epidermal growth factor receptor, EGFR), 人表皮生长因子受体2(human epidermal growth factor receptor 2, HER2, erbB-2/neu, ERBB2), 成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)发生共突变, 这些肺癌患者有望从SHP2抑制剂联合RTKs抑制剂的治疗中获益[16]。研究发现, SHP2抑制剂在体外和细胞内对致癌SHP2突变体的抑制作用与突变体自身的磷酸酶活性成反比[17, 18]。
SHP2参与肿瘤发生发展的多个信号通路的调控, 其中, SHP2是丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)信号通路完全激活的必要条件[2]。SHP2参与调控了RAS-RAF-MAPK信号通路(图1), SHP2作为RAS的直接激活剂, 通过促进下游配体的磷酸化而活化RAS, RAS过度激活又会引起癌症的发生, 因此, SHP2可作为治疗癌症潜在的靶点。SHP2可以去磷酸化KRAS, 增强了GTPase循环及与效应分子RAF的结合, 故抑制SHP2可以抑制KRAS-RAF-MAPK信号通路传导[19, 20]。Sprouty是RAS/ERK途径的负调节因子, 其磷酸化使其活性受到, 而SHP2可以促进Sprouty的去磷酸化, 阻碍了其与生长因子受体结合蛋白2(growth factor receptor-bound protein 2, grb2)结合, 使得Grb2-SOS复合物的形成, 激活MAPK下游的信号[21, 22]。此外, C-末端Src激酶(C-terminal Src kinase, Csk)与磷酸化的paxillin结合, 导致Src失活并抑制ERK的活化, 而激活的SHP2可以使paxillin蛋白去磷酸化, 增强EGF刺激的ERK1/2活化[23]。
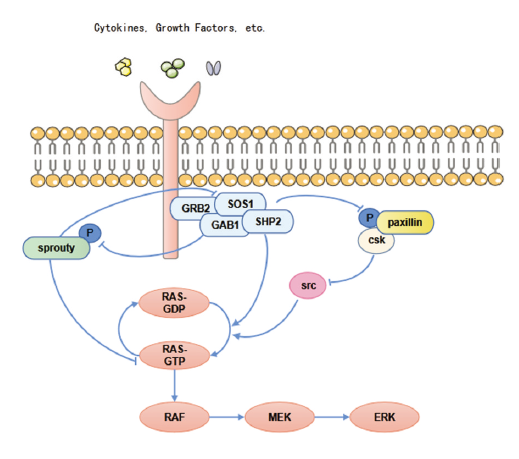 | 图1 SHP2调控RAS信号通路Fig.1 SHP2 regulates the RAS signaling pathway |
SHP2可以通过PTP催化活性依赖性的方式调节JAK/STAT通路, 使JAK2活化间接促进JAK/STAT介导的信号传导, 或是去磷酸化STAT直接抑制JAK/STAT通路活化[6, 24, 25]。SHP2与Yes关联蛋白1(Yes associated protein 1, YAP1)相互作用, 促进SHP2在细胞核内的积累, 激活了Wnt/β -catenin信号通路, 导致NSCLC患者的肿瘤进展以及不良预后[26]。有报道称NSCLC细胞中, KRAS和PI3K通路之间的串扰机制也是通过SHP2控制的[27]。SHP2可以通过PTP依赖性和非依赖性方式作为NF-κ B激活的双重调节因子, Src/Fn14/NF-kB轴也被证实在NSCLC的生长和转移中起着重要作用, 关于该轴的潜在作用也有待进一步的研究[28, 29]。
多项研究表明, SHP2抑制剂具有抗肿瘤血管生成的活性[30, 31]。SHP2抑制剂的应用使肿瘤血管内皮细胞ERK1/2磷酸化, 并激活STAT3磷酸化, 介导了抗肿瘤血管生成[32]。此外, 抑制SHP2可通过调控ASK1/c-Jun/SOX7轴, 降低小鼠肺恶性肿瘤病灶的微血管密度[33]。因此, 抑制SHP2也可以作为一种靶向肿瘤血管内皮细胞的新策略。
SHP2是少数已被证实的抑制细胞衰老的靶标之一。SHP2可以通过激活Src、FAK和MAPK来抑制衰老提升自我更新能力以促进乳腺癌的增殖, SHP2被抑制后, 衰老标志物p27和p53水平上升[34, 35]。在KRAS突变的NSCLC中, 抑制SHP2可以引起肿瘤细胞的衰老反应[36]。SHP2抑制剂的促肿瘤衰老作用有望运用于NSCLC的治疗中。
在肿瘤中单独使用SHP2抑制剂, 可以阻止MAPK通路的路径反馈激活及阻断RTK旁路激活, 有助于应对NSCLC的治疗耐药[37]。其单药的使用一直没有令人满意的临床试验数据披露, 但其拥有广阔的联合治疗潜力。
由于SHP2在致癌KRAS驱动的肿瘤中不可或缺的作用, 研究发现SHP2抑制剂与MEK抑制剂、HER2抑制剂、DOT1抑制剂、KRAS G12C抑制剂及PI3K抑制剂的联合使用都可以减退KRAS突变的某种或多种亚型的NSCLC肿瘤。SHP2是对MEK抑制剂产生耐药机制所必需的, 联合靶向SHP2和MEK时, 二者联用产生了协同效应, 可以使对MEK抑制剂产生耐药的KRAS突变型肿瘤恢复对MEK抑制剂的敏感性[36, 38]。HER2的拷贝数增加也是KRAS G12C突变NSCLC耐药的临床相关机制, 通过联合靶向抑制SHP2可以克服这种耐药机制[39]。此外, 上海药物所通过对KRAS突变型肿瘤进行蛋白磷酸化组学和药敏数据整合分析, 发现了联用DOT1抑制剂与SHP2抑制剂可以针对特定的KRAS突变肿瘤亚群进行有效治疗[33]。EMT是NSCLC对KRAS抑制剂sotorasib产生固有及获得性耐药的原因, 在这些EMT诱导的细胞中, PI3K被激活, 研究发现SHP2在MAPK通路的激活中起关键作用, 联合应用KRAS G12C抑制剂、PI3K抑制剂和SHP2抑制剂可使sotorasib获得性耐药小鼠肿瘤消退[40]。
EGFR突变是我国最为常见的NSCLC驱动基因变异, 目前酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI)耐药是EGFR抑制剂的治疗难点。相较于EGFR突变型, SHP2对EGFR野生型下游ERK磷酸化的促进作用要更加显著[41]。在EGFR抑制剂存在的情况下, SHP2的活性可以维持ERK的活性, 因此认为SHP2可能在肿瘤细胞耐药中扮演着重要角色[42]。联合使用SHP2与PI3K抑制剂可以抑制EGFRT790M/L858R双耐药突变细胞系H1975的增殖, 其抑制效应比其中任一单药都要更强。SHP2在奥希替尼耐药的肺腺癌(lung adenocarcinoma, LUAD)细胞中富集并促进肿瘤细胞的增殖, 敲除或抑制SHP2可以用于治疗EGFR抑制剂获得性耐药的NSCLC患者[43, 44]。SHP2低表达的LUAD细胞在体内外对奥希替尼的敏感性高于SHP2高表达的细胞, SHP2抑制剂可以通过阻断CXCL8-CXCR1/2环所介导的干性增强作用降低奥希替尼的耐药性[44]。联合抑制EGFR、SOS1和SHP2的疗法也在EGFR突变的NSCLC中具有治疗潜力[45]。
在ALK重排阳性的NSCLC患者中, 联合使用ALK TKI抑制剂和SHP2抑制剂, 可以有效抑制RAS和ERK1/2重新激活, 应对非ALK依赖性耐药[46]。Ⅲ 类BRAF突变属于激酶失活性异源二聚体, 多伴RAS激活突变与NF1缺失[47]。在Ⅲ 类BRAF突变细胞系中, MEK或RAF与SHP2的联合抑制表现出了显著的协同效应[48]。在MEK抑制剂tepotinib的获得性耐药发生中, MET以外的RTKs及下游信号元件的激活是其中关键一环。SHP2作为RTKs信号转导的下游效应因子, SHP2抑制剂的使用可以延缓和逆转MET扩增细胞对tepotinib的耐药性[49]。
免疫检查点抑制剂, 如抗细胞程序性死亡受体1(programmed cell death 1, PD-1)/细胞程序性死亡配体1(programmed cell death-ligand 1, PD-L1), 已成为治疗晚期NSCLC的有效药物。然而, 由于固有或获得性耐药性, 大约80%的患者对单独给予免疫治疗没有反应[50]。放射治疗(radiation therapy, XRT)可以克服部分患者的PD-1抑制剂耐药, 改善治疗结果, 但其疗效仍不理想[51]。研究发现, XRT在增加肿瘤PD-1表达的同时, 也会使巨噬细胞向M2型极化, 而SHP2抑制剂SHP099的加入可以逆转这一效应, 起到增强抗肿瘤免疫的作用[52]。
SHP2在不同治疗中作为生物标志物的潜力也在逐渐被挖掘。通过对公共数据库的分析, 与SHP2 mRNA低表达患者的总生存期相比, SHP2 mRNA高表达的NSCLC患者的总生存期和无进展生存期更差[44]。在免疫组化检查中, 有70%~94.1%的肺癌肿瘤组织可以表现出SHP2阳性, 与在癌旁与肿瘤间质中的表达有显著差异, 并与NSCLC的淋巴结转移密切相关[53, 54]。在接受免疫治疗与TKI治疗的肺癌患者中, SHP2的高表达与NSCLC患者的不良预后有关[53, 55, 56]。肿瘤组织中SHP2表达阴性的患者更容易从吉西他滨联合卡铂的化疗中获益[57]。
SHP2抑制剂在抗肿瘤治疗中可以起到重塑肿瘤免疫微环境的作用。载有SHP2抑制剂与CSF1R抑制剂的纳米颗粒可以将M2型肿瘤相关巨噬细胞极化为M1型, 并增加了肿瘤内T细胞和NK细胞的浸润, 从而增强了抗肿瘤免疫[58]。SHP2抑制剂与KRAS G12C抑制剂联用, 可以使CD8+ T细胞浸润增加, Treg细胞减少, 肿瘤相关的B细胞增加, 这可能是由于趋化因子CXCL9-11与CXCL13的分泌增加所介导的[31]。联合使用SHP2抑制剂与CXCR1/2抑制剂, 可以使因抑制RAS-RAF-ERK轴而激活的CXCR2信号通路被抑制, 促进抗肿瘤活性强的CD8+ T细胞的产生[59]。
早期SHP2抑制剂的开发主要为靶向PTP域催化口袋, 阻止酪氨酸磷酸化的底物进入催化位点, 主要代表有磺酸类抑制剂[60], 水杨酸类抑制剂[61]及天然来源的SHP2抑制剂[62]等。但由于PTP域高度保守的催化位点序列, 这类抑制剂缺乏特异性, 因此需要开发活性更高的SHP2抑制剂。
SHP2变构抑制剂的发现, 使得SHP2抑制剂真正进入了临床阶段。2016年诺华公司在《Nature》上报道了首个作用于SHP2变构位点的高选择性SHP2抑制剂SHP099, 该化合物可以结合到N-SH2、C-SH2和PTPT结构域之间的“ 隧道” 结合位点, 从而通过变构机制抑制SHP2的磷酸酶活性[63]。SHP2抑制剂RMC-4550不仅能将SHP2稳定于“ 自抑制状态” , 还可以通过降低SHP2在质膜上的丰度, 抑制SHP2发挥的磷酸酶功能[64]。诺华公司在SHP099的基础上开发了一种效用更好的变构抑制剂TNO155, 这也是首个进入临床试验的SHP2抑制剂[65]。之后诺华公司又发现在N端SHP2和PTP结构域界面形成的裂缝间有一个“ 闩样” 位点, 经过筛选后得到抑制剂SHP244。同时抑制“ 闩样” 结合位点和“ 隧道” 结合位点可以协同稳定SHP2的封闭非活性构象[66]。
靶向蛋白水解的嵌合体(proteolysis-targeting chimera, PROTAC)成为了近些年来小分子药物的研发热点, Wang等首次基于PROTAC技术设计开发了小分子SHP2抑制剂SHP2-D26, 证实靶向降解SHP2对于抑制SHP2活性是一种非常有效的策略[67]。靶向SHP2的PROTAC化合物SP4在HeLa细胞增殖抑制试验中显示, 其活性是SHP099的100倍[68]。目前暂无SHP2抑制剂上市, 已经进入临床试验的SHP2抑制剂多为变构抑制剂, 与NSCLC相关的SHP2抑制剂临床试验见表1。
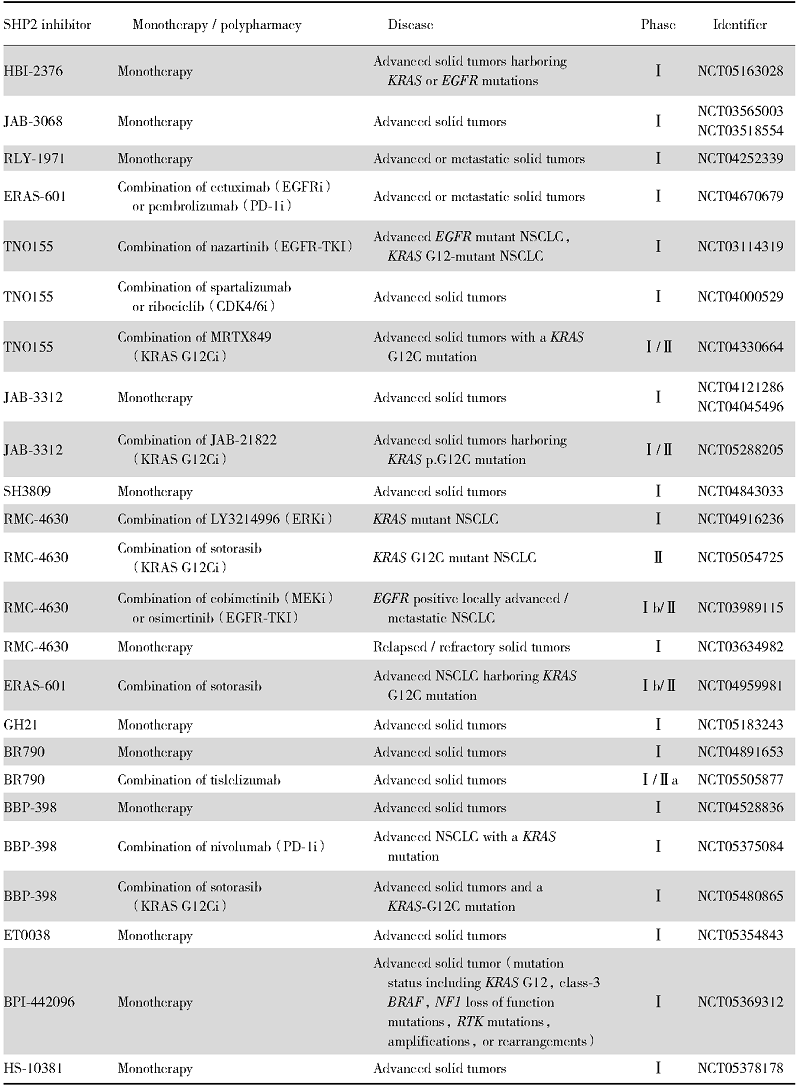
| 表1 针对实体瘤或非小细胞肺癌的SHP2抑制剂临床试验 Tab.1 Clinical trials of SHP2 inhibitors for solid tumors or NSCLC |
SHP2作为“ 分子开关” , 是肿瘤细胞与免疫细胞等细胞内多条信号通路的节点, 但一直以来都是难成药的靶点, 自SHP2的变构抑制剂SHP099面世后, 其潜力也变得备受瞩目。SHP2抑制剂对于多种癌症都显示出了治疗潜力, 多项关于NSCLC的临床前研究中也证实了抑制SHP2可以抑制肿瘤生长并促进抗肿瘤免疫。SHP2与NSCLC的多种分子靶向药物的耐药机制有关, 通过药物联用可以改善耐药, 重塑肿瘤的微环境, 但SHP2抑制剂的最佳联合应用策略还有待进一步的充分评估。此外, SHP2在肿瘤免疫微环境中的作用也是值得关注的话题, SHP2在不同的免疫细胞中可能存在差异性调控机制, 因此, 在肿瘤微环境的不同细胞中寻找SHP2新的底物与相互作用蛋白, 也许可以成为SHP2抑制剂新的治疗机会。总之, SHP2在NSCLC中是一个非常有潜力的靶点, 对于SHP2的相关机制探索需要进一步的研究。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|