

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
多组学联用在特异性败血症诊断中的价值
[麻硕1 , 林晓兰2 , 梁铭标2 , 谷祥拓2 , 钱鹏2 , 唐金陵1 , 梁会营1, 2  ]
]
]
|
|


麻硕(1995-),男,河北石家庄人,在读医学硕士研究生,主要研究方向为多组学数据处理、生物信息分析、机器学习。
目的 基于多组学数据,采用机器学习的分类算法,构建高性能的特异性致病菌感染败血症的早期诊断模型,并比较基于单组学和多组学数据模型的预测效果。方法 利用败血症患者早期血浆中测得的蛋白质组学数据、代谢组学数据以及多组学融合数据,使用支持向量机(support vector machine,SVM)算法分别构建三个诊断模型,对比三个模型的性能。结果 相较于单组学模型,利用多组学数据构建的模型效果最优,金黄色葡萄球菌感染组受试者工作特征曲线下的面积(area under the receiver operating characteristic curve,AUC)=0.97,非金黄色葡萄菌感染组AUC=0.94,非感染组AUC=0.94。结论 在特异性败血症早期诊断时,基于多组学相较于单组学构建的模型有较好的预测效果。
Objective A machine learning classification algorithm was employed to construct a high-performance early diagnosis model for sepsis infection with specific pathogenic bacteria based on multi-omics data. Then we compared the prediction effect between the single-omics model and the multi-omics model.Methods This was a secondary analysis of two observational studies. The omics data was extracted and integrated. Support vector machine (SVM) algorithm was used to construct three prediction models whose performance were compared mutually.Results The multi-omics model showed the best performance [( Staphylococcus aureus bacteria (SaB) vs. others, (area under the receiver operating characteristic curve, AUC)=0.97; non_SaB vs. others, AUC=0.94; Control vs. others, AUC=0.94] comparing with single-omic model.Conclusions Multi-omics prediction model had tremendous potential in identifying specific sepsis and performed better than single-omic model.
败血症是由感染引起的全身炎症状态, 这种感染与多器官衰竭的并发症会导致更致命的情况, 例如严重的败血症和感染性休克[1]。败血症是非心血管疾病引起重症监护室(intensive care unit, ICU)患者死亡的常见病因, 常见的致病菌有金黄色葡萄球菌(Staphylococcus aureus bacteria, SaB)、肺炎链球菌(Streptococcus pneumoniae, S.pneumoniae)、大肠埃希菌(Escherichia coli, E.coli)三种。其中由金黄色葡萄球菌感染引起的败血症往往存在较差的预后结局, 其总死亡率在20%~30%之间, 是ICU患者的严重疾病负担[2]。
因为败血症是一种全身性的、系统性的感染疾病, 任何特定生物标志物的单一数据都可能产生偏倚, 因此单独使用一种组学的信息都不能全面、无偏倚地反映败血症患者的复杂机体情况。多组学研究的出现为我们提供了一种从多维度、多视角去观察败血症的病理生理发生发展机制的方法。Langley等[1]基于一个训练集、两个验证集采用蛋白质组学和代谢组学的多组学技术, 发现了能够鉴别败血症和全身炎症反应综合征(systemic inflammatory response syndrome, SIRS)的高效能生物标志物。Wozniak[2]同样采用蛋白组学和代谢组学联用的多组学技术分析, 对200多例金黄色葡萄球菌败血症患者和正常人血清进行对比分析, 从而全面揭示早期预测和致病性特征, 为后续指导金黄色葡萄球菌败血症患者个性化治疗提供了有力工具。除了用于败血症治疗的高昂医疗保健费用外, 败血也会对患者本身及其护理照看人员产生很大影响。尽管每年都有大量关于如何降低与败血症相关的死亡率的研究, 但它仍然是全世界患者、临床医生和医疗系统面临的主要挑战, 因此早期识别败血症感染和改善患者预后是非常必要的[3]。目前, 大部分研究都聚焦在如何识别败血症与非败血症以及败血症不同预后之间的差异, 关于引起败血症的金黄色葡萄球菌与肺炎链球菌、大肠埃希菌之间的差异研究尚未看到。败血症的诊断主要依赖于实验室检查确定是否存在感染以及感染的类型, 然而实验室检测需要时间, 会导致治疗的滞后[4, 5]。研究表明不同致病菌引起的败血症死亡率存在差异, 且等血培养检测结果出具后再进行干预, 患者死亡率极高[6]。因此, 亟需开发一个可快速识别和鉴定败血症致病菌的方法, 实现精准地区分金黄色葡萄球菌与非金黄色葡萄球菌感染。
本文数据来源于两部分。第一部分来源于社区获得性肺炎和败血症结局诊断研究(Community Acquired Pneumonia and Sepsis Outcome Diagnostics, CAPSOD)(NCT00258869), 患者的纳入排除标准以及人口学特征在Langley等[1]的研究中有详细描述。第二部分来源于威斯康星大学麦迪逊分校健康中心有关金黄色葡萄球菌免疫反应的研究(#2018-0098)。关于患者的详细信息可见Wozniak等[2]发表的研究。在本次研究中, 从两组研究数据中提取了患者入院后用药前24小时内的血清检测数据, 包括197例金葡菌感染败血症、84例非金葡菌感染败血症、48例非败血症。
1.2.1 代谢物
第一部分数据, 使用无标记的液相气相色谱法-质谱法测定血清中的质荷比(mass-to-charge ratio, m/z)在100~1 000的生物化学分子。第二部分数据, 使用液相二级质谱法测定血清中的质荷比(m/z)在50~1 500的生物化学分子。通过取两部分数据的交集, 获得共表达的13个代谢物。
1.2.2 蛋白质
第一部分数据, 使用了特殊的方法对血清蛋白质进行测定和分析[1, 7, 8]。第二部分蛋白质数据的处理见Wozniak等[2]发表的研究。通过取两部分数据的交集, 获得共同表达的139个蛋白质。
采集到的数据统一按照Wozniak等[2]在研究中提到的方法处理数据, 将缺失值超过50%的变量删除, 然后使用归一化后的最小值来填充缺失值, 来满足两组数据的融合以及机器学习对数据格式的要求。
在单组学特征筛选时, 本研究使用了8种特征选择方法, 最后选择了效果较好且与支持向量机(support vector machine, SVM)预测模型相辅相成的SVM向前特征选择方法。其基本步骤分三步:第一步, 筛选包含信息丰富的变量; 第二步, 对变量赋予权重并从小到大排列; 第三步, 输出预测效果最好的5个特征变量。在多组学特征筛选时, 采用偏最小二乘判别分析(partial least squares discriminant analysis, PLS-DA)的筛选方法以及后融合的策略。
本文使用SVM机器学习方法构建模型。SVM是最有效的分类器之一, 可以在减少计算量的前提下处理更多属性或特征和防止模型过拟合[9]。数据样本按7∶3的比例随机划分为训练集和测试及验证集。模型参数随机初始化后, 以合页损失函数(hinge loss)作为损失函数, 使用线性核函数, 通过监督学习的方式, 进行五折交叉验证训练。本文使用了准确率、精确率、召回率、F1值以及受试者工作特征(receiver operating characteristic curve, ROC)曲线下面积(area under the ROC, AUC)评价模型的效。
在提取变量特征之前, 我们使用了主成分分析(principal component analysis, PCA)的无监督聚类方法, 来对高维的单组学数据进行降维重构。本文利用R软件(version 4.2.0)中mixOmics包中自带的PCA模块进行实验, 结果见图1。
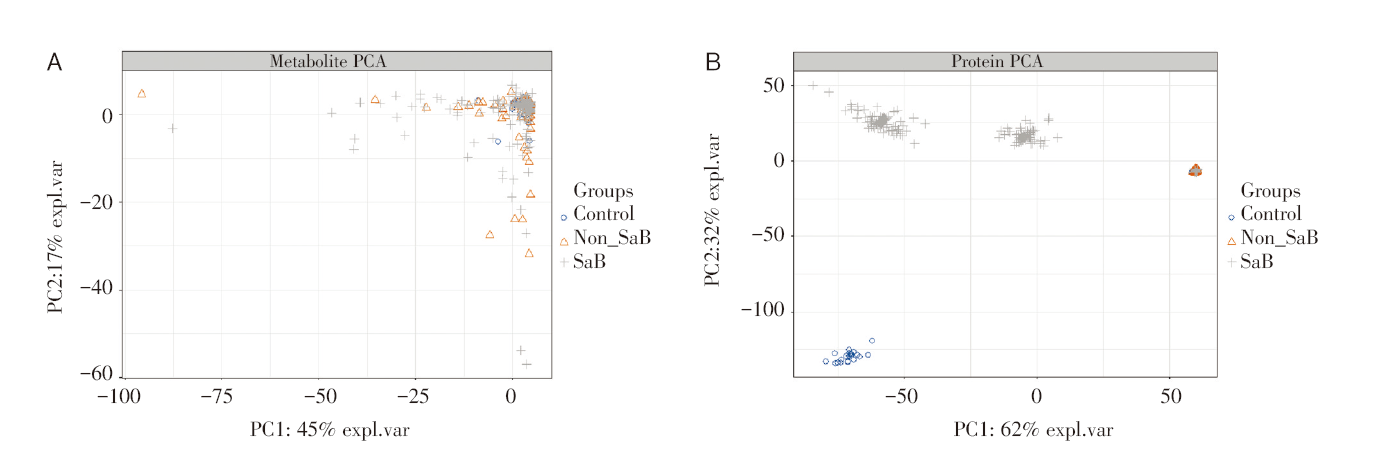 | 图1 PCA聚类 注: A. 代谢组数据的PCA聚类图; B. 蛋白质组数据PCA聚类图。Fig.1 PCA clustering Note: A. PCA cluster plot of metabolome data; B. PCA cluster plot of proteome data. |
本文尝试将感染金葡菌败血症的患者与感染其他菌种的败血症以及非败血症患者区分开。为充分利用特征信息, 本文使用了9种方法来对单组学建模数据进行了筛选, 每种方法筛选的前五个特征见表1。
| 表1 9种特征筛选方法得到的前五位特征 Tab.1 The top five features obtained by 9 feature screening methods |
本文使用的分类器模型是SVM模型。经过比较分析, 本文在单组学分析时选择了基于支持向量机的向前特征选择方法, 在多组学分析时选择了PLS-DA方法, 将筛选的排名前五位的特征纳入SVM模型。代谢组特征模型效果表现较差(AUC= 0.50±0.004, 准确率0.63, 精确率0.39, 召回率0.63, F1值0.48), 见图2A; 蛋白特征模型效果较好(AUC=0.89±0.006, 准确率0.87, 精确率0.91, 召回率0.87, F1值0.87), 见图2B; 代谢特征加蛋白特征的模型效果最好(AUC=0.95±0.000 2, 准确率0.86, 精确率0.88, 召回率0.86, F1值0.86), 见图2C。
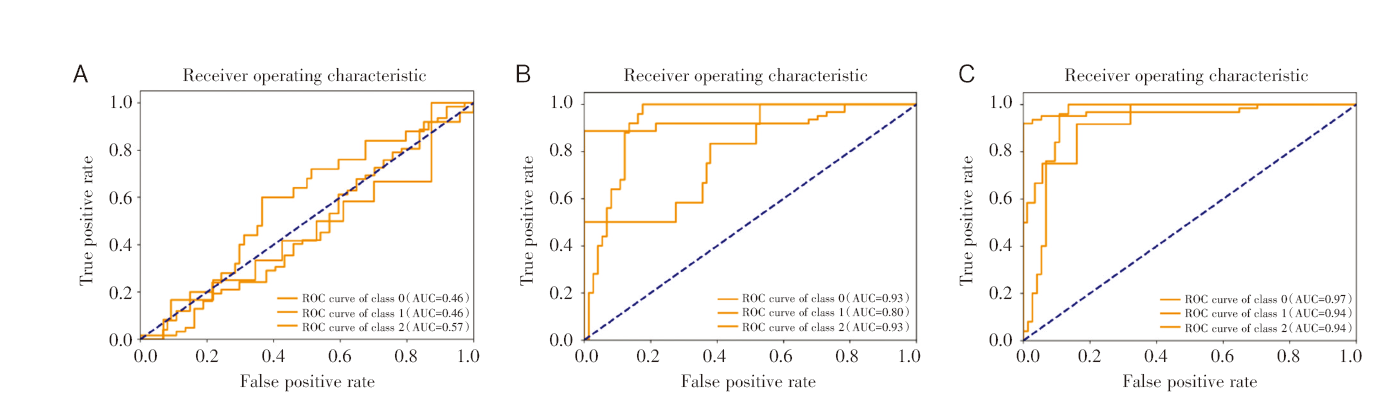 | 图2 SVM模型的ROC曲线 注:A. 5个代谢物特征模型的ROC曲线及AUC值; B. 5个蛋白质特征模型的ROC曲线及AUC值; C. 5个代谢物和5个蛋白质特征模型的ROC曲线及AUC值。0:金葡菌感染败血症组; 1:非感染组; 2:非金葡菌感染败血症组。Fig.2 ROC curve of SVM model Note: A. ROC curve and AUC value of 5 metabolite characteristics model; B. ROC curve and AUC value of 5 protein characteristics model; C. ROC curve and AUC value of 5 metabolite characteristics and protein characteristics model. 0: Staphylococcus aureus sepsis group; 1: non-infected group; 2: non-staphylococcus aureus sepsis group. |
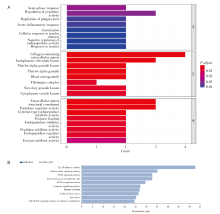
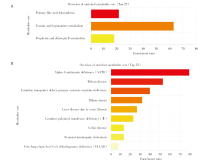
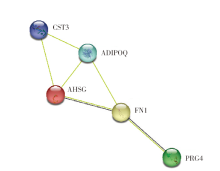
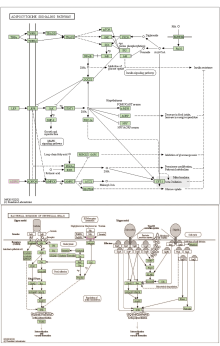
对多组学模型挑选出的蛋白质对应的基因进行基因本体(Gene Ontology, GO)术语富集分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)通路分析[10]。在生物学过程层面, 这些蛋白编码基因富集在肽酶调节、急性炎症反应、对胰岛素的反应等过程。在细胞成分层面, 基因富集在含胶原细胞外基质、内质网腔、血小板α颗粒管腔、血液微颗粒等物质成分, 见图3A。代谢通路富集分析显示, 5个蛋白编码基因与2型糖尿病、脂肪细胞因子信号通路、过氧化物酶体增殖物激活受体(peroxisome proliferator-activated receptors, PPAR)信号通路、细菌侵入上皮细胞通路相关, 见图3B。本文筛选的5个代谢物主要富集在(亚)牛磺酸和胆汁酸的合成上, 主要与α-1-抗胰蛋白酶缺乏症、威尔逊氏症、肉碱转运蛋白缺陷以及全身性肉碱缺乏症相关, 见图4。我们发现在蛋白质网络互相作用关系图中AHSG、FN1、PRG4关系密切且同时表达(见图5)[11], 而ADIPOQ[12]和FN1[13]基因分别在脂肪细胞因子信号通路和细菌侵入上皮细胞通路中扮演重要角色, 见图6(批准号230806)。
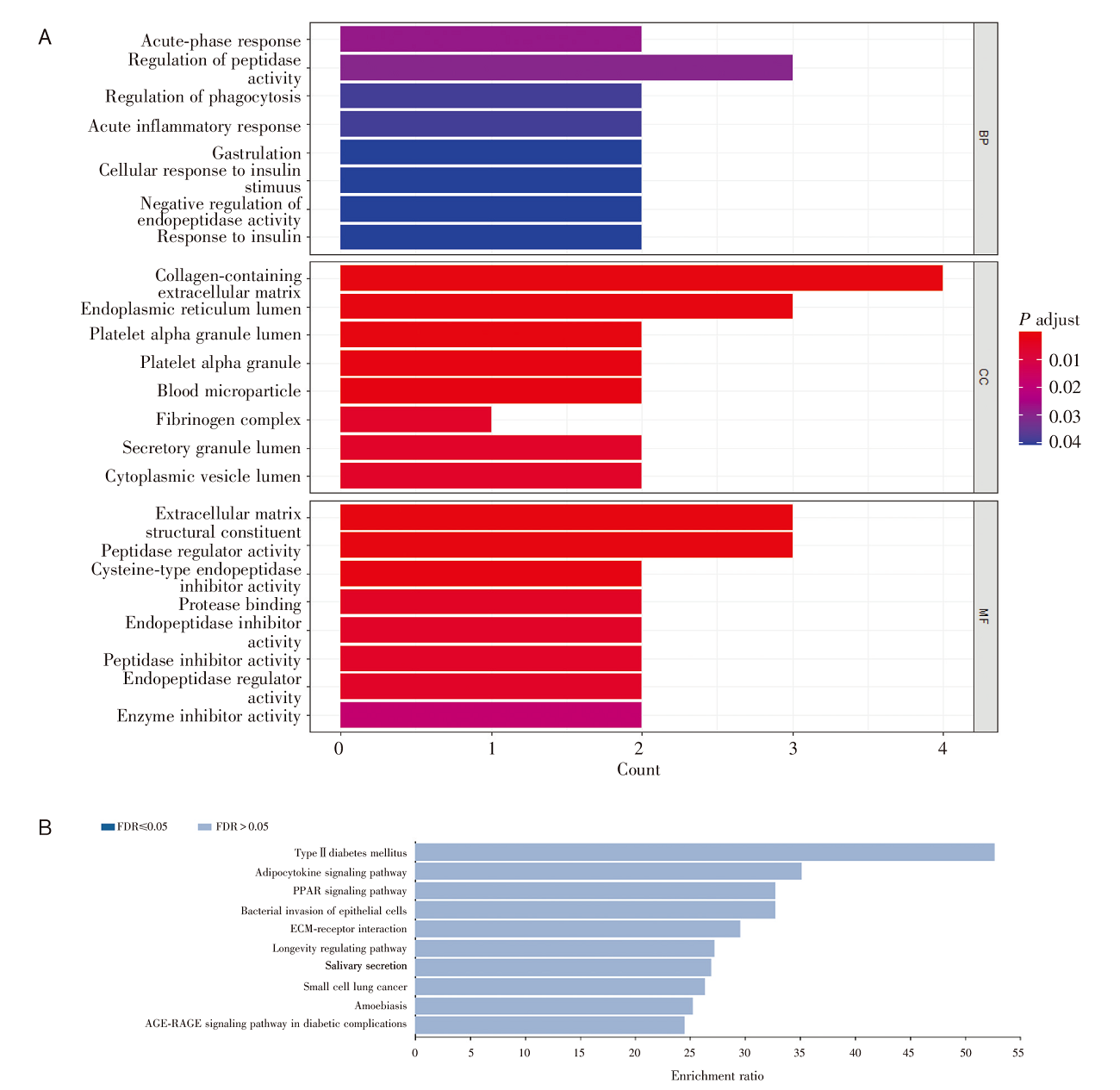 | 图3 基因富集分析 注:A. GO术语富集分析; BP, 生物学过程; CC, 细胞成分; MF, 分子功能; B. KEGG富集分析; FDR, 错误发现率。Fig.3 Gene enrichment analysis Note: A. GO terms enrichment analysis; BP, biological process; MCC, cellular components; F, molecular function; B. KEGG enrichment analysis; FDR, false discovery rate. |
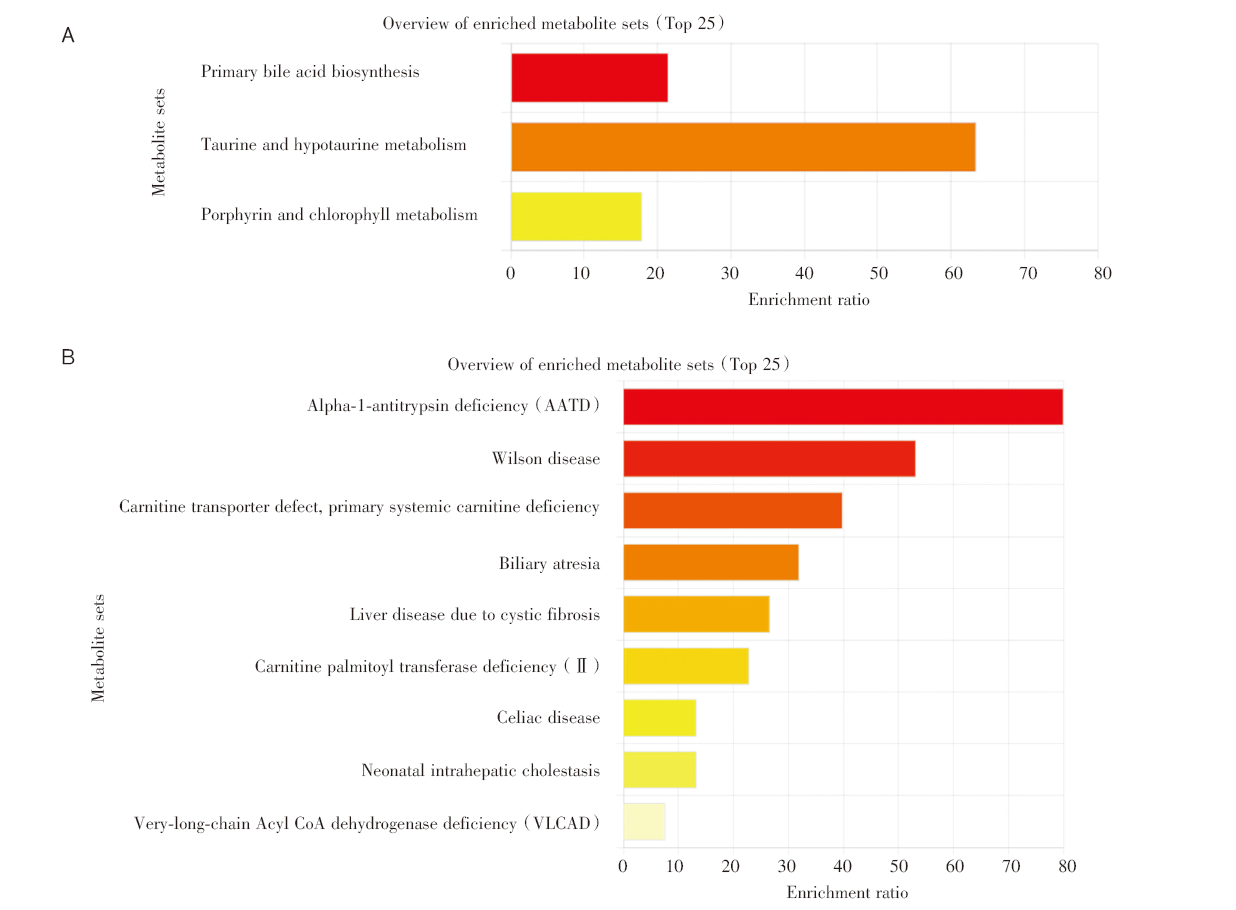 | 图4 代谢物富集分析 注:A. 代谢物功能富集分析; B. 代谢物疾病富集分析。Fig.4 Metabolite enrichment analysis Note: A. Metabolite functional enrichment analysis; B. Metabolite disease enrichment analysis. |
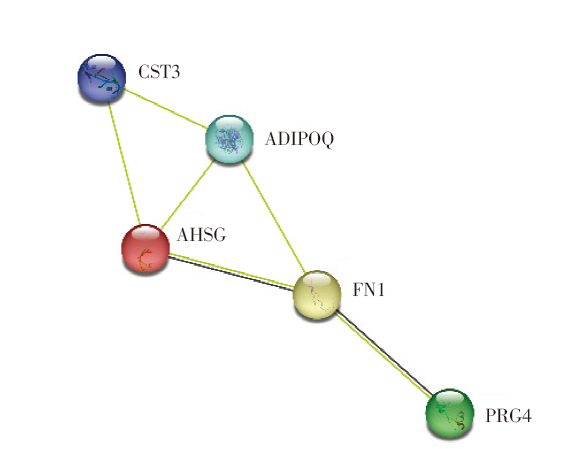 | 图5 蛋白质互相作用网络图Fig.5 Protein-protein interaction network diagram |
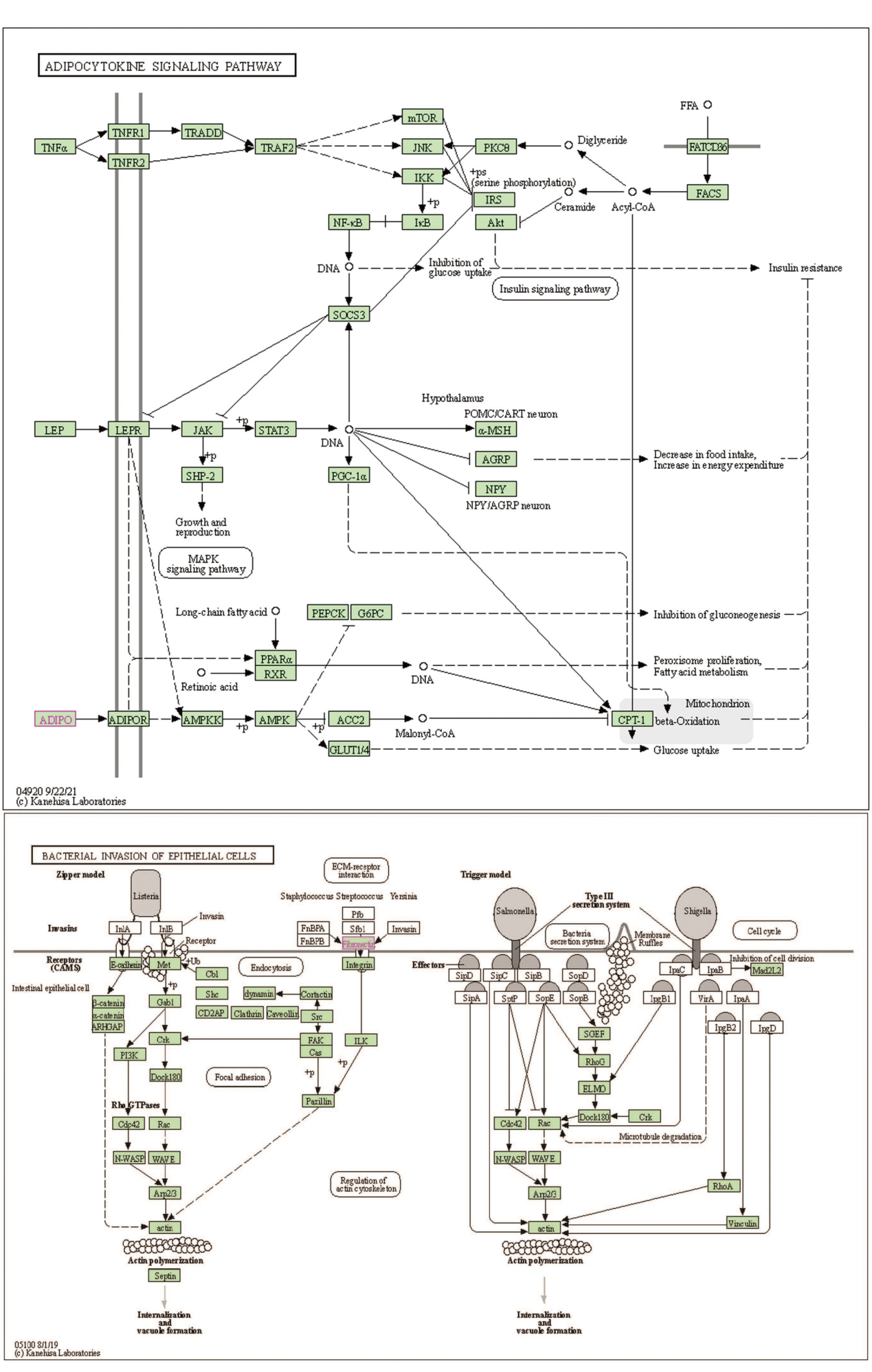 | 图6 ADIPOQ和FN1参与KEGG代谢通路图Fig.6 ADIPOQ and FN1 involved in KEGG metabolic pathway map |
本研究通过比较单组学和多组学模型的分类效果, 展示了多组学融合模型能充分利用数据特征进行高精度分类的性能, 为败血症患者的早期诊断提供可靠的支撑。本研究基于多组学的模型性能优于其他研究中基于电子病历的模型性能(AUC=0.79), 接近发病前4小时预测模型的性能(AUC=0.96)[14]。本研究基于多组学的SVM模型效果可以和集成SVM、随机森林(random forest, RF)、朴素贝叶斯(naive Bayesian, NB)、逻辑回归(logistic regression, LR)以及扩展梯度增强(eXtreme Gradient Boosting, XGBoost)的模型性能(AUC=0.96)持平[15]。相比于无监督方法(AUC = 0.82, F1值=0.65)[16], 本研究的模型性能依然具有优势。一种基于队列研究开发的早期预测败血症的人工智能算法结果显示模型的AUC为0.91[17], 也证明了本研究模型的良好预测能力。随着时间的推移, 败血症的定义已经从最初的细菌感染转变为炎症反应然后转变为现在的严重免疫和体内代谢功能障碍, 葡萄糖、游离脂肪酸等被认为是败血症新的模式分子[18]。临床研究还证明了脂肪酸代谢紊乱与败血症的全身炎症反应之间存在关联, 其中的饱和脂肪酸可以通过结合模式识别受体(pattern recognition receptors, PRR), 特别是Toll样受体(Toll-like receptors, TLRs)来诱导炎症[19]。同时我们筛选出来的肉碱类代谢物质也被认为参与了调节免疫反应与炎症的代谢通路[20]。以上结论从生物学过程发生的分子机制上证明了本文挑选的蛋白质和代谢物与败血症的发生密切相关, 也从侧面证明了多组学模型的可靠性。
但是本研究还是存在一些局限性。首先是样本量较小, 且研究的样本是从其他研究中获取的, 不具有随机性, 因此可能会引入偏倚。其次, 获取到的数据集中只包含13个可利用的代谢组学特征, 导致代谢组学模型性能不好。最后, 多组学模型的性能还需要在更大样本量的外部独立数据集中进行验证。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|